

Agrégation de peptides amyloïdes par des simulations numériques
Agrégation de peptides amyloïdes par des simulations numériques
Agrégation de peptides amyloïdes par des simulations numériques

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
32 3 Les petits pepti<strong>de</strong>s amyloï<strong>de</strong>s<br />
RMSD (Å)<br />
0 4 8<br />
H−bonds<br />
0 6 12<br />
RMSD (Å)<br />
0 4 8<br />
(a) (b)<br />
0 10 20<br />
Time (ns)<br />
H−bonds<br />
0 6 12<br />
(c) (d)<br />
0 10 20 30<br />
Time (ns)<br />
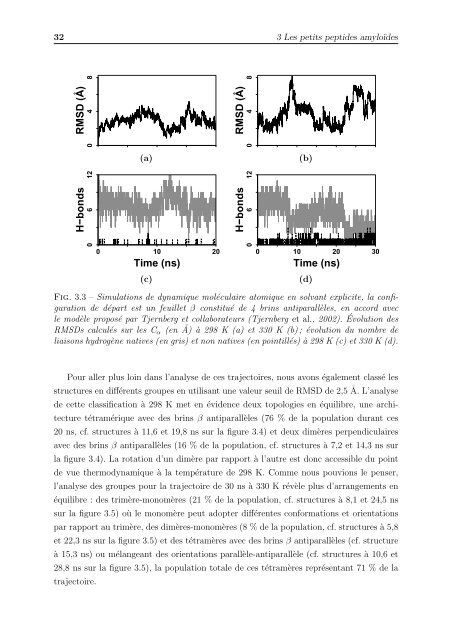
Fig. 3.3 – Simulations <strong>de</strong> dynamique moléculaire atomique en solvant explicite, la configuration<br />
<strong>de</strong> dé<strong>par</strong>t est un feuillet β constitué <strong>de</strong> 4 brins anti<strong>par</strong>allèles, en accord avec<br />
le modèle proposé <strong>par</strong> Tjernberg et collaborateurs (Tjernberg et al., 2002). Évolution <strong>de</strong>s<br />
RMSDs calculés sur les Cα (en ˚ A) à 298 K (a) et 330 K (b) ; évolution du nombre <strong>de</strong><br />
liaisons hydrogène natives (en gris) et non natives (en pointillés) à 298 K (c) et 330 K (d).<br />
Pour aller plus loin dans l’analyse <strong>de</strong> ces trajectoires, nous avons également classé les<br />
structures en différents groupes en utilisant une valeur seuil <strong>de</strong> RMSD <strong>de</strong> 2,5 ˚ A. L’analyse<br />
<strong>de</strong> cette classification à 298 K met en évi<strong>de</strong>nce <strong>de</strong>ux topologies en équilibre, une archi-<br />
tecture tétramérique avec <strong>de</strong>s brins β anti<strong>par</strong>allèles (76 % <strong>de</strong> la population durant ces<br />
20 ns, cf. structures à 11,6 et 19,8 ns sur la figure 3.4) et <strong>de</strong>ux dimères perpendiculaires<br />
avec <strong>de</strong>s brins β anti<strong>par</strong>allèles (16 % <strong>de</strong> la population, cf. structures à 7,2 et 14,3 ns sur<br />
la figure 3.4). La rotation d’un dimère <strong>par</strong> rapport à l’autre est donc accessible du point<br />
<strong>de</strong> vue thermodynamique à la température <strong>de</strong> 298 K. Comme nous pouvions le penser,<br />
l’analyse <strong>de</strong>s groupes pour la trajectoire <strong>de</strong> 30 ns à 330 K révèle plus d’arrangements en<br />
équilibre : <strong>de</strong>s trimère-monomères (21 % <strong>de</strong> la population, cf. structures à 8,1 et 24,5 ns<br />
sur la figure 3.5) où le monomère peut adopter différentes conformations et orientations<br />
<strong>par</strong> rapport au trimère, <strong>de</strong>s dimères-monomères (8 % <strong>de</strong> la population, cf. structures à 5,8<br />
et 22,3 ns sur la figure 3.5) et <strong>de</strong>s tétramères avec <strong>de</strong>s brins β anti<strong>par</strong>allèles (cf. structure<br />
à 15,3 ns) ou mélangeant <strong>de</strong>s orientations <strong>par</strong>allèle-anti<strong>par</strong>allèle (cf. structures à 10,6 et<br />
28,8 ns sur la figure 3.5), la population totale <strong>de</strong> ces tétramères représentant 71 % <strong>de</strong> la<br />
trajectoire.





