

Comparative day/night metatranscriptomic analysis of microbial ...
Comparative day/night metatranscriptomic analysis of microbial ...
Comparative day/night metatranscriptomic analysis of microbial ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Depth (m)<br />
0<br />
50<br />
100<br />
150<br />
200<br />
sequences, Proteobacteria were most abundant (41%;<br />
Fig. 3) and were dominated by a-Proteobacteria (22%),<br />
b-Proteobacteria (8%) and g-Proteobacteria (8%).<br />
Bacteroidetes (8%) and Firmicutes (12%, biased towards<br />
the <strong>day</strong> sample) were also well represented.<br />
Taxonomically binned mRNA sequences were compared<br />
with community composition data to ask whether<br />
taxa contributed to the HOT community mRNA in proportion<br />
to their representation in the <strong>microbial</strong> assemblage<br />
(i.e. whether taxa are equally transcriptionally active on a<br />
per-cell basis). Cyanobacteria dominated the transcript<br />
libraries (55% <strong>of</strong> sequences) with about tw<strong>of</strong>old higher<br />
representation than in the 16S rRNA amplicons or the cell<br />
count data (Fig. 3), indicating that there is more gene<br />
expression in these autotrophic bacterioplankton than in<br />
co-occurring heterotrophs (or possibly that their transcripts<br />
are longer-lived). When relative 16S rRNA abundance<br />
was calculated among just the heterotrophic<br />
groups (i.e. with cyanobacterial sequences removed),<br />
many taxa had similar contributions to the transcript pool<br />
and amplicon pool, suggesting comparable levels <strong>of</strong><br />
transcriptional activity on a per-gene basis within the limits<br />
<strong>of</strong> recognized biases <strong>of</strong> PCR amplification (Fig. 3).<br />
0 200 400 600<br />
chla (10 -3 μg l -1 )<br />
Prochlorococcus x 10 3 cells ml -1<br />
Synechococcus x 10 2 cells ml -1<br />
Nanoeukaryotes x 10 2 cells ml -1<br />
Heterotrophic bateria x 10 3 cells ml -1<br />
<strong>Comparative</strong> Metatranscriptomic Analysis 1361<br />
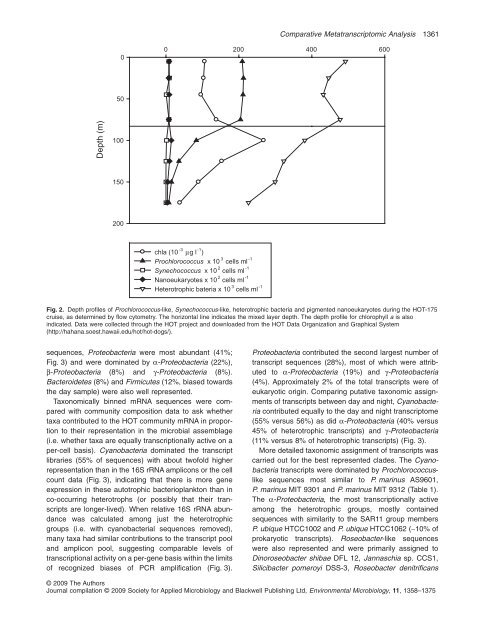
Fig. 2. Depth pr<strong>of</strong>iles <strong>of</strong> Prochlorococcus-like, Synechococcus-like, heterotrophic bacteria and pigmented nanoeukaryotes during the HOT-175<br />
cruise, as determined by flow cytometry. The horizontal line indicates the mixed layer depth. The depth pr<strong>of</strong>ile for chlorophyll a is also<br />
indicated. Data were collected through the HOT project and downloaded from the HOT Data Organization and Graphical System<br />
(http://hahana.soest.hawaii.edu/hot/hot-dogs/).<br />
Proteobacteria contributed the second largest number <strong>of</strong><br />
transcript sequences (28%), most <strong>of</strong> which were attributed<br />
to a-Proteobacteria (19%) and g-Proteobacteria<br />
(4%). Approximately 2% <strong>of</strong> the total transcripts were <strong>of</strong><br />
eukaryotic origin. Comparing putative taxonomic assignments<br />
<strong>of</strong> transcripts between <strong>day</strong> and <strong>night</strong>, Cyanobacteria<br />
contributed equally to the <strong>day</strong> and <strong>night</strong> transcriptome<br />
(55% versus 56%) as did a-Proteobacteria (40% versus<br />
45% <strong>of</strong> heterotrophic transcripts) and g-Proteobacteria<br />
(11% versus 8% <strong>of</strong> heterotrophic transcripts) (Fig. 3).<br />
More detailed taxonomic assignment <strong>of</strong> transcripts was<br />
carried out for the best represented clades. The Cyanobacteria<br />
transcripts were dominated by Prochlorococcuslike<br />
sequences most similar to P. marinus AS9601,<br />
P. marinus MIT 9301 and P. marinus MIT 9312 (Table 1).<br />
The a-Proteobacteria, the most transcriptionally active<br />
among the heterotrophic groups, mostly contained<br />
sequences with similarity to the SAR11 group members<br />
P. ubique HTCC1002 and P. ubique HTCC1062 (~10% <strong>of</strong><br />
prokaryotic transcripts). Roseobacter-like sequences<br />
were also represented and were primarily assigned to<br />
Dinoroseobacter shibae DFL 12, Jannaschia sp. CCS1,<br />
Silicibacter pomeroyi DSS-3, Roseobacter denitrificans<br />
© 2009 The Authors<br />
Journal compilation © 2009 Society for Applied Microbiology and Blackwell Publishing Ltd, Environmental Microbiology, 11, 1358–1375














