- Page 2:
Gene Function Analysis
- Page 6:
METHODS IN MOLECULAR BIOLOGYGene Fu
- Page 12:
PrefaceThis volume of Methods in Mo
- Page 16:
Prefaceixcolleagues demonstrate how
- Page 20:
xiiContentsPART III EXPERIMENTAL ME
- Page 26:
ICOMPUTATIONAL METHODS I
- Page 34:
4 BidautTable 1Input File Format Us
- Page 38:
6 BidautTable 2Folder Layout to Use
- Page 42:
8 Bidaut• alphaA: this is the num
- Page 46:
10 Bidautcomputing the maximum corr
- Page 50:
12 BidautFig. 3. The complete Clutr
- Page 54:
Table 3Some Identified Patterns (5,
- Page 58:
16 BidautFig. 4. This is a comparis
- Page 62: 18 BidautReferences1. Hughes, T. R.
- Page 66: 20 Kirov et al.way to associate gen
- Page 70: 22 Kirov et al.based on a study ass
- Page 74: 24 Kirov et al.1. Retrieve the gene
- Page 78: 26Fig. 1. Functional associations f
- Page 82: 28 Kirov et al.Fig. 2. Pathway anal
- Page 86: 30 Kirov et al.3. Gene symbols usag
- Page 90: 32 Kirov et al.9. OBO_Team, Open Bi
- Page 94: 3Estimating Gene Function With Leas
- Page 98: Estimating Gene Function With LS-NM
- Page 102: Estimating Gene Function With LS-NM
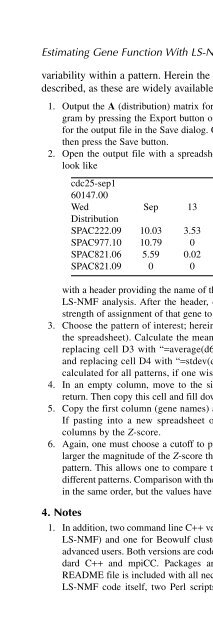
- Page 106: Estimating Gene Function With LS-NM
- Page 110: Estimating Gene Function With LS-NM
- Page 116: 46 Wang and Ochsresults posttreatme
- Page 120: 4From Promoter Analysis to Transcri
- Page 124: Prediction Using PAINT 51even in si
- Page 128: Prediction Using PAINT 53Fig. 1. A
- Page 132: Prediction Using PAINT 55first exon
- Page 138: 58 Gonye et al.Fig. 3. A network vi
- Page 142: 60 Gonye et al.exGeneList.txt) is a
- Page 146: 62 Gonye et al.(http://www.tm4.org)
- Page 150: 64 Gonye et al.does not span the en
- Page 154: 66 Gonye et al.4.7. Interpreting th
- Page 158: 68 Gonye et al.18. Dozmorov, M. G.,
- Page 162: 70 Uversky et al.in protein functio
- Page 166:
72 Uversky et al.sequence space and
- Page 170:
Table 1 (Continued)Server name URL
- Page 174:
76 Uversky et al.1.5. When to Use t
- Page 178:
78 Uversky et al.elucidating compos
- Page 182:
Table 2Averaged Frequencies of Diff
- Page 186:
82 Uversky et al.the box Raw Output
- Page 190:
84 Uversky et al.peculiarities of t
- Page 194:
86 Uversky et al.Fig. 4. Prediction
- Page 198:
88 Uversky et al.13. Uversky, V. N.
- Page 202:
90 Uversky et al.50. Vucetic, S., O
- Page 206:
92 Uversky et al.84. Ritter, L. M.,
- Page 210:
6Sybil: Methods and Software for Mu
- Page 214:
Sybil: Multiple Genome Comparison a
- Page 218:
Sybil: Multiple Genome Comparison a
- Page 222:
Sybil: Multiple Genome Comparison a
- Page 226:
Sybil: Multiple Genome Comparison a
- Page 230:
Sybil: Multiple Genome Comparison a
- Page 234:
Sybil: Multiple Genome Comparison a
- Page 238:
Sybil: Multiple Genome Comparison a
- Page 242:
7Estimating Protein Function Using
- Page 246:
Estimating Protein Function Using P
- Page 250:
Estimating Protein Function Using P
- Page 254:
Estimating Protein Function Using P
- Page 258:
Estimating Protein Function Using P
- Page 262:
Estimating Protein Function Using P
- Page 266:
Estimating Protein Function Using P
- Page 270:
Estimating Protein Function Using P
- Page 274:
Estimating Protein Function Using P
- Page 278:
Estimating Protein Function Using P
- Page 282:
130 Davuluriinteracting proteins an
- Page 286:
Table 1Web URLs of Promoter, TF Dat
- Page 290:
134 DavuluriPWM-based models do not
- Page 294:
136 DavuluriTF-map alignments of or
- Page 298:
138 Davuluridiscussed which program
- Page 302:
140 DavuluriTable 2ER-a-Responsive
- Page 306:
Table 3Sample Data Matrix Represent
- Page 310:
Table 3 (Continued)Class MYCMAX MYC
- Page 314:
146 DavuluriFig. 3. (A) CART Tree:
- Page 318:
148 Davuluri11. Vlieghe, D., Sandel
- Page 322:
150 Davuluri44. Berezikov, E., Gury
- Page 326:
9Mining Biomedical Data Using MetaM
- Page 330:
Mining Biomedical Data Using MMTx a
- Page 334:
Mining Biomedical Data Using MMTx a
- Page 338:
Mining Biomedical Data Using MMTx a
- Page 342:
Mining Biomedical Data Using MMTx a
- Page 346:
Mining Biomedical Data Using MMTx a
- Page 350:
Mining Biomedical Data Using MMTx a
- Page 354:
Mining Biomedical Data Using MMTx a
- Page 358:
Mining Biomedical Data Using MMTx a
- Page 362:
172 Ho et al.Fig. 1. Artificial exa
- Page 366:
174 Ho et al.allowing for cases whe
- Page 370:
176 Ho et al.A different measure is
- Page 374:
178 Ho et al.3.1.3. LA and Generali
- Page 378:
180 Ho et al.The ECF-statistic can
- Page 382:
182 Ho et al.In the special case of
- Page 386:
184 Ho et al.Fig. 5. An illustratio
- Page 390:
186 Ho et al.Fig. 7. The power curv
- Page 394:
188 Ho et al.this section were not
- Page 398:
190 Ho et al.References1. Schena, M
- Page 402:
IIIEXPERIMENTAL METHODS
- Page 406:
194 Caldwell et al.for sequences th
- Page 410:
196 Caldwell et al.query because it
- Page 414:
198 Caldwell et al.Fig. 1. (A) Prot
- Page 418:
200 Caldwell et al.outside primer o
- Page 422:
202 Caldwell et al.5. Targeting scr
- Page 426:
204 Caldwell et al.will allow the s
- Page 430:
206 Caldwell et al.3.1.6. Plasmid P
- Page 434:
208 Caldwell et al.PCR amplify the
- Page 438:
210 Caldwell et al.8. Thawing cells
- Page 442:
212 Zhang et al.Going one step beyo
- Page 446:
214 Zhang et al.Fig. 2. Generation
- Page 450:
216 Zhang et al.Perform PCR cycles,
- Page 454:
218 Zhang et al.Fig. 4. Schematic m
- Page 458:
220 Zhang et al.Fig. 5. Replacement
- Page 462:
13Construction of Simple and Effici
- Page 466:
DNA Vector-Based shRNA-Expression S
- Page 470:
DNA Vector-Based shRNA-Expression S
- Page 474:
DNA Vector-Based shRNA-Expression S
- Page 478:
DNA Vector-Based shRNA-Expression S
- Page 482:
DNA Vector-Based shRNA-Expression S
- Page 486:
DNA Vector-Based shRNA-Expression S
- Page 490:
DNA Vector-Based shRNA-Expression S
- Page 494:
DNA Vector-Based shRNA-Expression S
- Page 498:
DNA Vector-Based shRNA-Expression S
- Page 502:
244 Hust et al.overcome by two appr
- Page 506:
246 Hust et al.Fig. 1. Schematic de
- Page 510:
248 Hust et al.interaction during p
- Page 514:
250 Hust et al.3.4. Titering1. Inoc
- Page 518:
252 Hust et al.10. Shortly before u
- Page 522:
254 Hust et al.activity by preservi
- Page 526:
15A Bacterial/Yeast Merged Two-Hybr
- Page 530:
Screening in Yeast With a Bacterial
- Page 534:
Screening in Yeast With a Bacterial
- Page 538:
Screening in Yeast With a Bacterial
- Page 542:
Screening in Yeast With a Bacterial
- Page 546:
Screening in Yeast With a Bacterial
- Page 550:
Screening in Yeast With a Bacterial
- Page 554:
Screening in Yeast With a Bacterial
- Page 558:
Screening in Yeast With a Bacterial
- Page 562:
Screening in Yeast With a Bacterial
- Page 566:
Screening in Yeast With a Bacterial
- Page 570:
Screening in Yeast With a Bacterial
- Page 574:
Screening in Yeast With a Bacterial
- Page 578:
Screening in Yeast With a Bacterial
- Page 582:
Screening in Yeast With a Bacterial
- Page 586:
Screening in Yeast With a Bacterial
- Page 590:
Screening in Yeast With a Bacterial
- Page 594:
16A Bacterial/Yeast Merged Two-Hybr
- Page 598:
Dual Bait-Compatible Bacterial Two-
- Page 602:
Dual Bait-Compatible Bacterial Two-
- Page 606:
Dual Bait-Compatible Bacterial Two-
- Page 610:
Dual Bait-Compatible Bacterial Two-
- Page 614:
Dual Bait-Compatible Bacterial Two-
- Page 618:
Dual Bait-Compatible Bacterial Two-
- Page 622:
Dual Bait-Compatible Bacterial Two-
- Page 626:
Dual Bait-Compatible Bacterial Two-
- Page 630:
Dual Bait-Compatible Bacterial Two-
- Page 634:
Dual Bait-Compatible Bacterial Two-
- Page 638:
Dual Bait-Compatible Bacterial Two-
- Page 642:
Dual Bait-Compatible Bacterial Two-
- Page 646:
318 Thibodeau-Beganny and Joungbeen
- Page 650:
320 Thibodeau-Beganny and JoungFig.
- Page 654:
322 Thibodeau-Beganny and JoungFig.
- Page 658:
324 Thibodeau-Beganny and JoungTypi
- Page 662:
326 Thibodeau-Beganny and JoungFig.
- Page 666:
328 Thibodeau-Beganny and JoungPCR
- Page 670:
330 Thibodeau-Beganny and Joung16-1
- Page 674:
332 Thibodeau-Beganny and Joung2. P
- Page 678:
334 Thibodeau-Beganny and Joung11.
- Page 682:
336 IndexKknockin (gene knockin) 19