

Research Group - IPK Gatersleben
Research Group - IPK Gatersleben
Research Group - IPK Gatersleben

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
Abteilung Genbank/Department of Genebank<br />
In the context of the map-based isolation of the GA-insensitive<br />
semi-dwarfing gene sdw3, the target interval,<br />
comprising 0.04 cM on a high resolution genetic map of<br />
barley, has been further analysed by comparative mapping<br />
in rice and Brachypodium sylvaticum. As to the latter,<br />
a contiguous sequence of 365 kb was obtained from<br />
screening a BAC library and sequence analysis of an overlapping<br />
set of four BACs. The target interval in rice encompasses<br />
180 kb and comprises 4 genes of which 15 are<br />
not present in Brachypodium. The corresponding region<br />
in Brachypodium sylvaticum measures about 0 kb with<br />
16 annotated genes of which seven are not present in<br />
rice. The remaining nine genes are conserved between<br />
both species and show perfect collinearity. Screening of<br />
a barley BAC library by using eight genes from the target<br />
interval led to the identification of seven BAC contigs. Of<br />
these, only two contigs identified by two genes spaced<br />
by 800 bp in Brachypodium showed overlaps. Based on<br />
survey sequencing of one representative BAC per barley<br />
contig, 11 complete and two fragmented genes were<br />
identified. Seven genes were found in a colinear position<br />
in rice while four genes are unique to barley. The results<br />
confirm previous observations on varying gene repertoires<br />
of orthologous regions in rice, Brachypodium and<br />
Triticeae and provide further proof of the necessity to rely<br />
on several genomes when exploiting synteny with model<br />
genomes for map-based cloning. Based on the sequence<br />
information, three candidate genes for GA-signalling are<br />
further investigated by re-sequencing sdw3 mutant and<br />
wild type genotypes in barley and by analysing induced<br />
mutants identified from a TILLING population (V.T.H.<br />
Giang).<br />
To assist the functional verification of candidate genes<br />
a TILLING (targeted induced local lesions in genomes)<br />
population is being developed. The establishment of an<br />
M mutant population consisting of 10,49 independent<br />
plants was completed. For systematic screening of this<br />
population, DNA of 8,500 M mutants has been arranged<br />
in 8-fold D pools. During the reference period, screening<br />
of a subset comprising 7,348 lines was completed for<br />
five genes. The averaged mutation frequency amounts to<br />
1/500kb. 35 % of the mutations revealed missense alleles<br />
and 4 % resulted in truncations. 35 % of the silent mutations<br />
were present in noncoding regions (S. Gottwald).<br />
As a step towards the development of genome-based<br />
strategies for the utilisation of genetic diversity a study<br />
on linkage disequilibrium (LD) mapping in barley has<br />
been completed. To this end, a world-wide collection of<br />
5 spring barley cultivars was phenotyped in multi-location<br />
trials for five agronomic traits (heading date, plant<br />
height, thousand grain weight, starch content, protein<br />
content). Single Nucleotide Polymorphisms (SNPs) in nine<br />
candidate genes were correlated with trait data in order<br />
to identify associations. Using a mixed model that includes<br />
information on population structure, row-type and origin<br />
of the accessions, significant marker trait associations<br />
34<br />
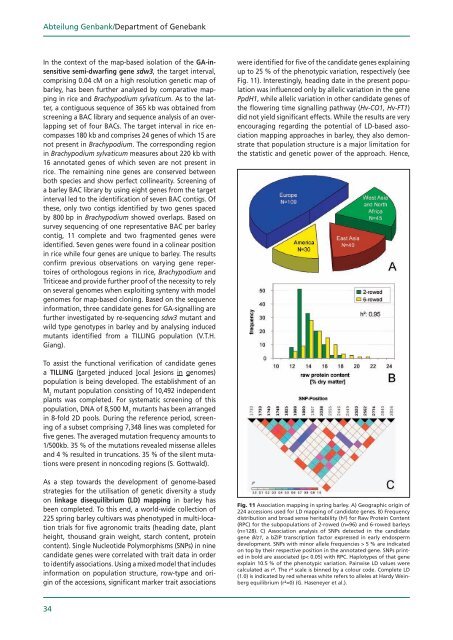
were identified for five of the candidate genes explaining<br />
up to 5 % of the phenotypic variation, respectively (see<br />
Fig. 11). Interestingly, heading date in the present population<br />
was influenced only by allelic variation in the gene<br />
PpdH1, while allelic variation in other candidate genes of<br />
the flowering time signalling pathway (Hv-CO1, Hv-FT1)<br />
did not yield significant effects. While the results are very<br />
encouraging regarding the potential of LD-based association<br />
mapping approaches in barley, they also demonstrate<br />
that population structure is a major limitation for<br />
the statistic and genetic power of the approach. Hence,<br />
Fig. 11 Association mapping in spring barley. A) Geographic origin of<br />
4 accessions used for LD mapping of candidate genes. B) Frequency<br />
distribution and broad sense heritability (h²) for Raw Protein Content<br />
(RPC) for the subpopulations of -rowed (n=96) and 6-rowed barleys<br />
(n=1 8). C) Association analysis of SNPs detected in the candidate<br />
gene Blz1, a bZIP transcription factor expressed in early endosperm<br />
development. SNPs with minor allele frequencies > 5 % are indicated<br />
on top by their respective position in the annotated gene. SNPs printed<br />
in bold are associated (p< 0.05) with RPC. Haplotypes of that gene<br />
explain 10.5 % of the phenotypic variation. Pairwise LD values were<br />
calculated as r². The r² scale is binned by a colour code. Complete LD<br />
(1.0) is indicated by red whereas white refers to alleles at Hardy Weinberg<br />
equilibrium (r²=0) (G. Haseneyer et al.).










