Genetische Krankheiten - DocCheck Campus
Genetische Krankheiten - DocCheck Campus
Genetische Krankheiten - DocCheck Campus

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
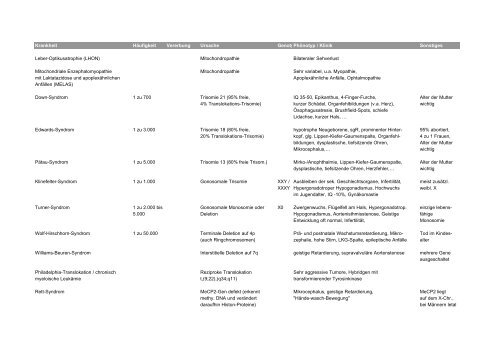
Krankheit Häufigkeit Vererbung Ursache GenotypPhönotyp / Klinik Sonstiges<br />
Leber-Optikusatrophie (LHON) Mitochondropathie Bilateraler Sehverlust<br />
Mitochondriale Enzephalomyopathie Mitochondropathie Sehr variabel, u.a. Myopathie,<br />
mit Laktatazidose und apoplexähnlichen Apoplexähnliche Anfälle, Ophtalmopathie<br />
Anfällen (MELAS)<br />
Down-Syndrom 1 zu 700 Trisomie 21 (95% freie, IQ 35-50, Epikanthus, 4-Finger-Furche, Alter der Mutter<br />
4% Translokations-Trisomie) kurzer Schädel, Organfehlbildungen (v.a. Herz), wichtig<br />
Ösophagusatresie, Brushfield-Spots, schiefe<br />
Lidachse, kurzer Hals, …<br />
Edwards-Syndrom 1 zu 3.000 Trisomie 18 (80% freie, hypotrophe Neugeborene, sgR, prominenter Hinter- 95% abortiert,<br />
20% Translokations-Trisomie) kopf, glg. Lippen-Kiefer-Gaumenspalte, Organfehl- 4 zu 1 Frauen,<br />
bildungen, dysplastische, tiefsitzende Ohren, Alter der Mutter<br />
Mikrocephalus,… wichtig<br />
Pätau-Syndrom 1 zu 5.000 Trisomie 13 (80% freie Trisom.) Mirko-/Anophthalmie, Lippen-Kiefer-Gaumenspalte, Alter der Mutter<br />
dysplastische, tiefsitzende Ohren, Herzfehler,… wichtig<br />
Klinefelter-Syndrom 1 zu 1.000 Gonosomale Trisomie XXY / Ausbleiben der sek. Geschlechtsorgane, Infertilität, meist zusätzl.<br />
XXXY Hypergonadotroper Hypogonadismus, Hochwuchs weibl. X<br />
im Jugendalter, IQ -10%, Gynäkomastie<br />
Turner-Syndrom 1 zu 2.000 bis Gonosomale Monosomie oder X0 Zwergenwuchs, Flügelfell am Hals, Hypergonadotrop. einzige lebens-<br />
5.000 Deletion Hypogonadismus, Aortenisthmisstenose, Geistige fähige<br />
Entwicklung oft normal, Infertilität, Monosomie<br />
Wolf-Hirschhorn-Syndrom 1 zu 50.000 Terminale Deletion auf 4p Prä- und postnatale Wachstumsretardierung, Mikro- Tod im Kindes-<br />
(auch Ringchromosomen) zephalie, hohe Stirn, LKG-Spalte, epileptische Anfälle alter<br />
Williams-Beuren-Syndrom Interstitielle Deletion auf 7q geistige Retardierung, supravalvuläre Aortenstenose mehrere Gene<br />
ausgeschaltet<br />
Philadelphia-Translokation / chronisch Reziproke Translokation Sehr aggressive Tumore, Hybridgen mit<br />
myeloische Leukämie t,(9;22),(q34;q11) transformierender Tyrosinkinase<br />
Rett-Syndrom MeCP2-Gen defekt (erkennt Mikrocephalus, geistige Retardierung, MeCP2 liegt<br />
methy. DNA und verändert "Hände-wasch-Bewegung" auf dem X-Chr.,<br />
daraufhin Histon-Proteine) bei Männern letal
Krankheit Häufigkeit Vererbung Ursache GenotypPhönotyp / Klinik Sonstiges<br />
Prader-Willi-Syndrom 1 zu 16.000 75% Deletion auf 15q(beim Mann) Hypotrophe Babys, Fresssucht -> Adipositas, aktives männl.<br />
20% maternale Disomie / Muskelhypotonie, Entwicklungsverzögerung, geistige PWS-Gen<br />
Imprinting-Fehler Retardierung, auffällige Mund- und Augenpartie deletiert<br />
Angelman-Syndrom / Happy-Puppet-Syn. 65% Deletion auf 15q (bei Frau) schwere geistige Retardierung, unkontrollierte, aktives weibl.<br />
6% Imprinting-Störung ataktische Bewegungen, Lachanfälle, Krampfleiden AS-Gen deletiert<br />
3% paternale Disomie<br />
Marfan-Syndrom aut.-dom. Fibrillin-Gen defekt Fehlerhafte Kollagensynthese, hohe Penetr.,<br />
lange und schmale Extremitäten, Arachnodaktylie, Unterschiedl.<br />
Linsenluxation, Aortenaneurisma, Trichter-/Hühner- Expressivität,<br />
brust, Sternbergzeichen (Daumen ragt aus geschl. Beta-Blocker-<br />
Faust), Hochwuchs Therapie<br />
Neurofibrimatose (M. von Recklinghausen) 1 zu 3.000 aut.-dom. 50% Neumutation Café-au-lait-Flecken, "Sommersprossen", Unterschiedl.<br />
Neurofibrome (Knoten aus periph. Nerven und Expressivität<br />
Bindegewebe), überaktives ras-G-Protein (wirkt auf<br />
MAPK,...)<br />
Cystische Fibrose / Mukoviszidose 1 zu 2.000 aut.-rez. häufig F508-Mutation (Deletion Hochvisköses Sekret muköser Drüsen, pulmonale häufigste a.-rez<br />
von Phe) -> defektes CFTR- Symptome (chronische obstruktive Lungenerkrankung, Erkrankung,<br />
Protein (=Chloridkanal); Fehler Ateminsuffizienz), intestinale Symptome (Pankreas- HZF 1:20<br />
am Spleiß-Akzeptor insuffizienz->Malabsorption->Hypoproteinämie, Mekoniumileus,<br />
Rektumprolaps), erhöhte Na + - und Cl - -Ausscheidung<br />
im Schweiß, atrophierte Vas deferens<br />
Phenylketonurie (PKU) 1 zu 10.000 aut.-rez. Über 70 Mutationen -> Defekt schwere geisteige Retardierung, epileptische Anfälle, HZF 1:50,<br />
der Phenylalaninhydroxylase -> Übererregbarkeit, Pigementstörungen, Urin riecht wie Therapie durch<br />
[Phe] steigt; Phe als Teratogen Mäuseurin Phe-arme Diät<br />
durch maternale PKU v.a. bei Kindern<br />
und Schwang.,<br />
heterogene Exp.<br />
Rachitis x-chr.-dom. Störung der tubulären Hypophosphatämie, Skelettveränderungen, O-Beine, durch Komp.<br />
Rückresorption von Phospat Minderwuchs milderer Verlauf<br />
bei Frauen<br />
Incontinetia pigmenti 1 zu 40.000 x-chr.-dom. Bläschen-> Hyperkeratosestreifen -> im 30. LJ sym- bei männl. Feten<br />
tomfrei (nur noch Zellen mit gesundem aktiven X) letal
Krankheit Häufigkeit Vererbung Ursache GenotypPhönotyp / Klinik Sonstiges<br />
Duchenne-Muskel-Dystrophie (DMD) 1 zu 3.000 x-chr.-rez. Große Frame-shift-Mutation im erst ab 3. LJ: Ungeschicklichkeit, Fallneigung, Pseudo- Konduktorinnen<br />
(bei Männern) Dystrophin-Gen -> keinerlei Hypertrophie der Waden, Schwierigkeiten beim haben leichte<br />
Dystrophin (1/3 Neumutationen) Aufstehen (Gower-Zeichen); ab 10. LJ: Rollstuhl, Symptomatik,<br />
Muskelatrophie -> Ateminsuffizienz, LWS-Hyperlordose Tod unter 20<br />
Becker-Muskel-Dystrophie x-chr.-rez. keine Frame-Shift-Mutation leichtere und verzögerte DMD-Symptomatik Tod zw. 40-50<br />
Hämophilie A / B x-chr.-rez. 40% Geninversion, viele Gerinnungsstörung-> Hämatomneigung, Gelenkblut- Therapie durch<br />
Misssensemutationen ungen in Knie- und Sprunggelenken Subst. des<br />
Gerinnungsfakt.<br />
Rot-Grün-Blindheit x-chr.-rez.<br />
Roberts-Syndrom aut.-rez. gleiche Symptomatik wie Contergan-Geschädigte Bsp. Für<br />
(Tetrafokomelie, LKG-Spalte,…) Phänokopie<br />
Holt-Oram-Syndrom aut.-dom. ähnliche Symptomatik wie Contergan-Geschädigte Bsp. Für<br />
aber nur obere Extremität betroffen Phänokopie<br />
Chorea Huntington 1 zu 10.000 aut.-dom. expandierendes CAG-Repeat unwillkürliche Bewegungen, psychische Störungen, Manifestation im<br />
(nur 1% Neumutationen) Demenz; Symptome von Generation zu Generation Erw.alter, Gen.<br />
gravierender und früher manifestiert, Antizipation Imp.: männliches<br />
Gen -><br />
schellerer Verl.<br />
Fragiles-X-Syndrom / Martin-Bell-Syndrom 1 zu 4.000 x-chr.-rez. fragile terminale Stelle an qX geistige Retardierung, äußere Auffälligkeiten (große nur 80% der<br />
(bei Männern) Ohren, langes, schmales Gesicht, Progenie, …), männl. Anlage-<br />
Verhaltens- und Sprachentwicklungsstörungen täger erkrankt<br />
Sichelzellanämie kodominant / Transversionssubstitution -> erhöhte Blut-Viskosität -> verstopfte Kapillaren (-> Heterozygote<br />
aut.-rez. verformte Erythrozyten Milzinfarkt, Pneumonie), verstärkter Ery-Abbau -> klinisch normal<br />
hämolytische Anämie<br />
Klumpfuß 1 zu 1.000 Multifaktorielle Erkrankung Bsp.für<br />
Schwellenwert
Krankheit Häufigkeit Vererbung Ursache GenotypPhönotyp / Klinik Sonstiges<br />
Xeroderma pigmentosa aut.-rez. Mutation eines XP-Proteins Extreme UV-Lichtempfindlichkeit -> trockene, pigmentder<br />
Nukleotidexzisionsreperatur ierte Haut, hohes Hautkrebsrisiko, Melanome, Leukämie<br />
erblicher Brustkrebs aut.-dom. Ausfälle in Homologer Mamma- und Ovarialkarzinome Verursacht 5%<br />
Rekombination (v.a. Mutation aller Mammader<br />
BRCA1/2-Gene (TF)) karzinome<br />
Hereditäre kolorektale Karzinome ohne aut.-dom. Mutation der Missmatch-Repa- früh auftretende kolorektale Karzinome, Neoplasien,<br />
Polyposis (HNPCC) ratur-Gene (oder Basenexz.- Mikrsatelliteninstabilität<br />
reparatur-Gene)<br />
Cockayne-Syndrom aut.-rez. Ausfall in transkriptions- erhöhte Apoptose-Rate -> geistige Retardierung, DNA-Reperatur<br />
gekoppelter DNA-Reparatur Minderwuchs, Infektanfälligkeit, Gesichtsdysmorphien und Transkript.<br />
betroffen<br />
Nijmegen-Breakage-Syndrom aut.-rez. Defekt der Doppelstrang-Repa- prä- und postnatale Wachstumsretardierung, Mikroration<br />
zephalie, Gesichtsdysmorphien, mentale Retardierung,<br />
Immundefekte