news and views© 2010 Nature America, Inc. All rights reserved.criteria for crop improvement (Fig. 1). Therecent papers 1–7 will guide selection of parentsby clarifying the substantial differencesin active gene contents between differentmaize inbreds. Assuming that a major basisof heterosis is the complementation of thesedifferences in inbreds 1,2 , the new informationprovides pointers as to which parents shouldcomplement well in heterotic hybrids.Interestingly, the genome-wide expressionstudies of Swanson-Wagner et al. 5 suggestthat there is much differential gene expressionbetween alleles in hybrids relative to theinbred parents and that this is driven primarilyby the paternal allele of trans-actingexpression quantitative trait loci—genes thatexert their effect by regulating the expressionof other genes, often elsewhere in thegenome. This provocative finding raises thequestion of the extent to which heterosisdepends also on epigenetic and parent-oforiginimprinting effects, and of whetherbreeders should therefore focus on these loci,in particular when making crosses in specificdirections.The positions and frequencies of recombinationevents relative to the positions ofdiverse alleles influence tremendously theefficiencies of improved plant productionbecause they determine how readily newcombinations of alleles are created. Goreet al. 4 revealed that some 21% of genes liein low-recombination regions around thecentromeres and, thus, that variation inthese genes is difficult to exploit in breeding,except by means of new chromosomecombinations and hybrids. Vielle-Calzadaet al. 3 defined >100 regions characterizedby low genetic diversity among maize lines.These regions may therefore be associatedwith traits that contributed to domestication(such as a larger number of kernels thatremain attached to the cob and lack a stonycasing) and consequently have been selectedThe wealth of information inthese reports should acceleratebreeding projects aimed atgenerating superior varieties ofcorn and other crops.in all modern maize breeding programs.Other regions are highly polymorphic andmay therefore be associated with adaptationto different geographic regions and sourcesof variation that breeders select and elaborate.The largest maize-breeding companieshave probably already sequenced many corngenomes, or parts of them, and they havemuch more accurate information on phenotypesof lines in a wide array of environmentsthan the public sector does. Nevertheless,the studies 1–7 will provide these companieswith much new data and analyses that willbe used to drive their breeding programs.Unfortunately, most smaller companies lackthe resources (e.g., databases and informationtechnology systems) to fully exploit the newgenomic information, suggesting that the gapbetween those who have this capability andthose who do not will continue to widen.Those who stand to benefit especially fromthese reports 1–7 are the breeders in the publicsector and small companies that are seekingto provide improved lines for poorer societies.Foremost among them is the ConsultativeGroup on International AgriculturalResearch and its tropical maize breedingefforts spearheaded by the InternationalMaize and Wheat Improvement Center(Centro Internacional de Mejoramientode Maíz y Trigo; CIMMYT). CIMMYT hasstruggled to fully embrace genomics andhas lagged behind the leading private sectorcompanies in exploiting genomics inits breeding program, on which so manydepend. Let us hope that these publicationswill prove sufficiently compelling to inspiretheir government funders and other publicsectorbreeders to expedite the application ofgenomics in crop breeding.COMPETING INTERESTS STATEMENTThe author declares no competing financial interests.1. Schnable, P.S. et al. Science 326, 1112–1115(2009).2. Springer, N.M. et al. PLoS Genet. 5, e1000734(2009).3. Vielle-Calzada, J.-P. et al. Science 326, 1078 (2009).4. Gore, M.A. et al. Science 326, 1115–1117 (2009).5. Swanson-Wagner, R.A. et al. Science 326, 1118–1120 (2009).6. McMullen, M.D. et al. Science 325, 737–740(2009).7. Buckler, E.S. et al. Science 325, 714–718(2009).Spilling the beans on legume biologySoybean is the most recentaddition to the rapidly growinglist of crops for which a highqualitydraft genome is nowavailable. Writing in Nature,Schmutz et al. 1 report that the1.1-gigabasesoybeangenome—thelargest shotgunsequencedplantgenome—ispredictedto encode46,000 genes.Two genomeduplicationevents are likelyRoy Kaltschmidtto account for the observationthat ~75% of these genesare found in multiple copies.Although the importanceof soybean as a source ofprotein and oil alone testifiesto the potential implicationsof understanding its geneticmakeup, this genome willalso serve as the reference for~20,000 leguminous speciesthat play a critical ecologicalrole through their unique abilityto fix nitrogen with the help ofrhizobial bacteria. Availability ofthe genome should acceleratethe association of quantitativetrait loci of nutritional,economic and ecologicallyimportant traits with the causalDNA sequences from soybeanin the near future. In the longerterm, the genome will likelyalso be leveraged to improvethe way in which a range ofleguminous subsistence cropsare used to both replenish soilnitrogen through crop rotationand meet the expanding needsof developing nations forprotein and energy.Peter Hare1. Schmutz, J. et al. Nature 463, 178–183 (2010).nature biotechnology volume 28 number 2 february 2010 145
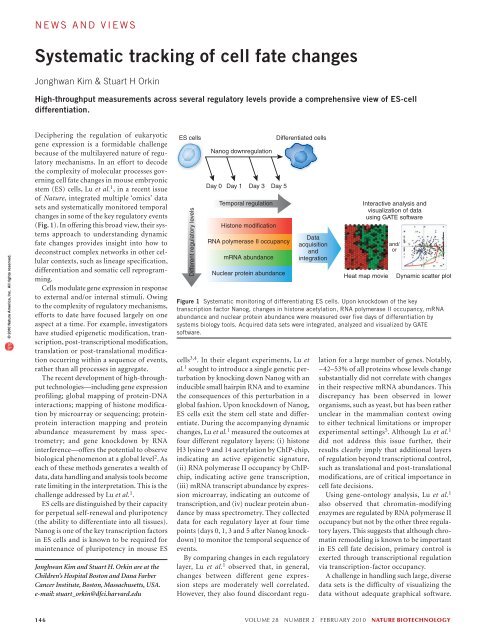
news and viewsSystematic tracking of cell fate changesJonghwan Kim & Stuart H OrkinHigh-throughput measurements across several regulatory levels provide a comprehensive view of ES-celldifferentiation.© 2010 Nature America, Inc. All rights reserved.Deciphering the regulation of eukaryoticgene expression is a formidable challengebecause of the multilayered nature of regulatorymechanisms. In an effort to decodethe complexity of molecular processes governingcell fate changes in mouse embryonicstem (ES) cells, Lu et al. 1 , in a recent issueof Nature, integrated multiple ‘omics’ datasets and systematically monitored temporalchanges in some of the key regulatory events(Fig. 1). In offering this broad view, their systemsapproach to understanding dynamicfate changes provides insight into how todeconstruct complex networks in other cellularcontexts, such as lineage specification,differentiation and somatic cell reprogramming.Cells modulate gene expression in responseto external and/or internal stimuli. Owingto the complexity of regulatory mechanisms,efforts to date have focused largely on oneaspect at a time. For example, investigatorshave studied epigenetic modification, transcription,post-transcriptional modification,translation or post-translational modificationoccurring within a sequence of events,rather than all processes in aggregate.The recent development of high-throughputtechnologies—including gene expressionprofiling; global mapping of protein-DNAinteractions; mapping of histone modificationby microarray or sequencing; proteinproteininteraction mapping and proteinabundance measurement by mass spectrometry;and gene knockdown by RNAinterference—offers the potential to observebiological phenomenon at a global level 2 . Aseach of these methods generates a wealth ofdata, data handling and analysis tools becomerate limiting in the interpretation. This is thechallenge addressed by Lu et al. 1 .ES cells are distinguished by their capacityfor perpetual self-renewal and pluripotency(the ability to differentiate into all tissues).Nanog is one of the key transcription factorsin ES cells and is known to be required formaintenance of pluripotency in mouse ESJonghwan Kim and Stuart H. Orkin are at theChildren’s Hospital Boston and Dana FarberCancer Institute, Boston, Massachusetts, USA.e-mail: stuart_orkin@dfci.harvard.eduES cellsDifferent regulatory levelsNanog downregulationDay 0 Day 1Day 3Temporal regulationHistone modificationRNA polymerase II occupancymRNA abundanceDifferentiated cellsDay 5Nuclear protein abundanceFigure 1 Systematic monitoring of differentiating ES cells. Upon knockdown of the keytranscription factor Nanog, changes in histone acetylation, RNA polymerase II occupancy, mRNAabundance and nuclear protein abundance were measured over five days of differentiation bysystems biology tools. Acquired data sets were integrated, analyzed and visualized by GATEsoftware.cells 3,4 . In their elegant experiments, Lu etal. 1 sought to introduce a single genetic perturbationby knocking down Nanog with aninducible small hairpin RNA and to examinethe consequences of this perturbation in aglobal fashion. Upon knockdown of Nanog,ES cells exit the stem cell state and differentiate.During the accompanying dynamicchanges, Lu et al. 1 measured the outcomes atfour different regulatory layers: (i) histoneH3 lysine 9 and 14 acetylation by ChIP-chip,indicating an active epigenetic signature,(ii) RNA polymerase II occupancy by ChIPchip,indicating active gene transcription,(iii) mRNA transcript abundance by expressionmicroarray, indicating an outcome oftranscription, and (iv) nuclear protein abundanceby mass spectrometry. They collecteddata for each regulatory layer at four timepoints (days 0, 1, 3 and 5 after Nanog knockdown)to monitor the temporal sequence ofevents.By comparing changes in each regulatorylayer, Lu et al. 1 observed that, in general,changes between different gene expressionsteps are moderately well correlated.However, they also found discordant regu-DataacquisitionandintegrationInteractive analysis andvisualization of datausing GATE softwareHeat map movieand/orDynamic scatter plotlation for a large number of genes. Notably,~42–53% of all proteins whose levels changesubstantially did not correlate with changesin their respective mRNA abundances. Thisdiscrepancy has been observed in lowerorganisms, such as yeast, but has been ratherunclear in the mammalian context owingto either technical limitations or improperexperimental settings 5 . Although Lu et al. 1did not address this issue further, theirresults clearly imply that additional layersof regulation beyond transcriptional control,such as translational and post-translationalmodifications, are of critical importance incell fate decisions.Using gene-ontology analysis, Lu et al. 1also observed that chromatin-modifyingenzymes are regulated by RNA polymerase IIoccupancy but not by the other three regulatorylayers. This suggests that although chromatinremodeling is known to be importantin ES cell fate decision, primary control isexerted through transcriptional regulationvia transcription-factor occupancy.A challenge in handling such large, diversedata sets is the difficulty of visualizing thedata without adequate graphical software.146 volume 28 number 2 february 2010 nature biotechnology
- Page 3 and 4: volume 28 number 2 february 2010COM
- Page 5 and 6: in this issue© 2010 Nature America
- Page 7 and 8: © 2010 Nature America, Inc. All ri
- Page 10 and 11: NEWS© 2010 Nature America, Inc. Al
- Page 12 and 13: NEWS© 2010 Nature America, Inc. Al
- Page 14 and 15: NEWS© 2010 Nature America, Inc. Al
- Page 16 and 17: © 2010 Nature America, Inc. All ri
- Page 18 and 19: © 2010 Nature America, Inc. All ri
- Page 20 and 21: © 2010 Nature America, Inc. All ri
- Page 22 and 23: NEWS feature© 2010 Nature America,
- Page 24 and 25: uilding a businessComing to termsDa
- Page 26 and 27: uilding a business© 2010 Nature Am
- Page 28 and 29: correspondence© 2010 Nature Americ
- Page 30 and 31: correspondence© 2010 Nature Americ
- Page 32 and 33: correspondence© 2010 Nature Americ
- Page 34 and 35: correspondence© 2010 Nature Americ
- Page 36 and 37: case studyNever againcommentaryChri
- Page 38 and 39: COMMENTARY© 2010 Nature America, I
- Page 40 and 41: COMMENTARY© 2010 Nature America, I
- Page 42 and 43: patents© 2010 Nature America, Inc.
- Page 44 and 45: patents© 2010 Nature America, Inc.
- Page 46 and 47: news and viewsChIPs and regulatory
- Page 48 and 49: news and viewsFrom genomics to crop
- Page 52 and 53: news and views© 2010 Nature Americ
- Page 54 and 55: e s o u r c eRational association o
- Page 56 and 57: e s o u r c e© 2010 Nature America
- Page 58 and 59: e s o u r c e© 2010 Nature America
- Page 60 and 61: e s o u r c e© 2010 Nature America
- Page 62 and 63: © 2010 Nature America, Inc. All ri
- Page 64 and 65: B r i e f c o m m u n i c at i o n
- Page 66 and 67: i e f c o m m u n i c at i o n sAUT
- Page 68 and 69: lettersa1.5 kb hVPrIntron 112.5 kbA
- Page 70 and 71: letters© 2010 Nature America, Inc.
- Page 72 and 73: letters© 2010 Nature America, Inc.
- Page 74 and 75: l e t t e r sReal-time imaging of h
- Page 76 and 77: l e t t e r sFigure 2 Time-lapse li
- Page 78 and 79: l e t t e r s© 2010 Nature America
- Page 80 and 81: l e t t e r sRational design of cat
- Page 82 and 83: l e t t e r s© 2010 Nature America
- Page 84 and 85: l e t t e r s© 2010 Nature America
- Page 86 and 87: sample fluorescence was measured as
- Page 88 and 89: careers and recruitmentFourth quart