

California Biomedical Industry - California Healthcare Institute
California Biomedical Industry - California Healthcare Institute
California Biomedical Industry - California Healthcare Institute

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
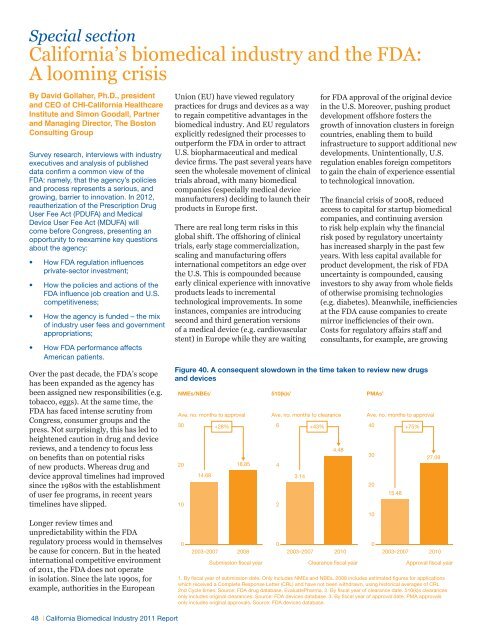
Special section<strong>California</strong>’s biomedical industry and the FDA:A looming crisisBy David Gollaher, Ph.D., presidentand CEO of CHI-<strong>California</strong> <strong>Healthcare</strong><strong>Institute</strong> and Simon Goodall, Partnerand Managing Director, The BostonConsulting GroupSurvey research, interviews with industryexecutives and analysis of publisheddata confirm a common view of theFDA: namely, that the agency’s policiesand process represents a serious, andgrowing, barrier to innovation. In 2012,reautherization of the Prescription DrugUser Fee Act (PDUFA) and MedicalDevice User Fee Act (MDUFA) willcome before Congress, presenting anopportunity to reexamine key questionsabout the agency:• How FDA regulation influencesprivate-sector investment;• How the policies and actions of theFDA influence job creation and U.S.competitiveness;• How the agency is funded – the mixof industry user fees and governmentappropriations;• How FDA performance affectsAmerican patients.Over the past decade, the FDA’s scopehas been expanded as the agency hasbeen assigned new responsibilities (e.g.tobacco, eggs). At the same time, theFDA has faced intense scrutiny fromCongress, consumer groups and thepress. Not surprisingly, this has led toheightened caution in drug and devicereviews, and a tendency to focus lesson benefits than on potential risksof new products. Whereas drug anddevice approval timelines had improvedsince the 1980s with the establishmentof user fee programs, in recent yearstimelines have slipped.Longer review times andunpredictability within the FDAregulatory process would in themselvesbe cause for concern. But in the heatedinternational competitive environmentof 2011, the FDA does not operatein isolation. Since the late 1990s, forexample, authorities in the European48 | <strong>California</strong> <strong>Biomedical</strong> <strong>Industry</strong> 2011 ReportUnion (EU) have viewed regulatorypractices for drugs and devices as a wayto regain competitive advantages in thebiomedical industry. And EU regulatorsexplicitly redesigned their processes tooutperform the FDA in order to attractU.S. biopharmaceutical and medicaldevice firms. The past several years haveseen the wholesale movement of clinicaltrials abroad, with many biomedicalcompanies (especially medical devicemanufacturers) deciding to launch theirproducts in Europe first.There are real long term risks in thisglobal shift. The offshoring of clinicaltrials, early stage commercialization,scaling and manufacturing offersinternational competitors an edge overthe U.S. This is compounded becauseearly clinical experience with innovativeproducts leads to incrementaltechnological improvements. In someinstances, companies are introducingsecond and third generation versionsof a medical device (e.g. cardiovascularstent) in Europe while they are waitingfor FDA approval of the original devicein the U.S. Moreover, pushing productdevelopment offshore fosters thegrowth of innovation clusters in foreigncountries, enabling them to buildinfrastructure to support additional newdevelopments. Unintentionally, U.S.regulation enables foreign competitorsto gain the chain of experience essentialto technological innovation.The financial crisis of 2008, reducedaccess to capital for startup biomedicalcompanies, and continuing aversionto risk help explain why the financialrisk posed by regulatory uncertaintyhas increased sharply in the past fewyears. With less capital available forproduct development, the risk of FDAuncertainty is compounded, causinginvestors to shy away from whole fieldsof otherwise promising technologies(e.g. diabetes). Meanwhile, inefficienciesat the FDA cause companies to createmirror inefficiencies of their own.Costs for regulatory affairs staff andconsultants, for example, are growingFigure …And a 40. consequent A consequent slowdown in slowdown the time taken in to the review time new taken drugs to and review devicesnew drugsand devicesNMEs/NBEs 1Ave. no. months to approval302010014.682003–2007+28%18.852008Submission fiscal year510(k)s 2Ave. no. months to clearance64203.142003–2007+43%Clearance fiscal year1. By fiscal year of submission date. Only includes NMEs and NBEs. 2008 includes estimated figures for applicationswhich received a Complete Response Letter (CRL) and have not been withdrawn, using historical averages of CRL2nd Cycle times. Source: FDA drug database, EvaluatePharma. 2. By fiscal year of clearance date. 510(k)s clearancesonly includes original clearances. Source: FDA devices database. 3. By fiscal year of approval date. PMA approvalsonly includes original approvals. Source: FDA devices database.4.482010PMAs 3Ave. no. months to approval40302010015.482003–2007+75%27.082010Approval fiscal year



