

European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Cytogenetics<br />
to reports from Saudi Arabia and Kuwait but the frequency of abnormalities<br />
varied with a mixture of +8, t(8;21) & t(15;17). Morphologically<br />
our patients differed with M2 subtype as the commonest, whereas,<br />
their reports showed M4 and M3 subtypes as most frequently occurring<br />
subtype in their patient population respectively.<br />
P0290. Screening of subtle copy number changes in Aicardi<br />
Syndrome Patients with a high resolution X-chromosome array-<br />
CGH<br />
S. Yilmaz 1 , H. Fontaine 1 , K. Brochet 1 , M. Grégoire 1 , M. Devignes 2 , J. Schaff 1 , C.<br />
Philippe 1 , C. Nemos 1 , J. McGregor 3 , P. Jonveaux 1 ;<br />
1 University hospital of Nancy, Vandoeuvre les Nancy, France, 2 CNRS-UMR<br />
7503, LORIA, Vandoeuvre les Nancy, France, 3 INSERM Unité 689, Paris,<br />
France.<br />
Aicardi syndrome is a very uncommon neurodevelopmental disorder<br />
affecting almost exclusively females. Chief features include infantile<br />
spasms, corpus callosal agenesis, and chorioretinal abnormalities.<br />
Aicardi syndrome is a sporadic disorder and hypothesized to be caused<br />
by heterozygous mutations in an X linked-gene but up to now without<br />
any defined candidate region on the X chromosome. Array based comparative<br />
genomic hybridisation has become the method of choice for<br />
the detection of microdeletions and microduplications at high resolution.<br />
In this study, for the first time, 18 Aicardi syndrome patients were<br />
analyzed with a full-coverage X-chromosomal BAC arrays at a theoretical<br />
resolution of 82 kb. Copy number changes were validated by real<br />
time quantitation. No disease-associated aberrations were identified.<br />
For such conditions as Aicardi syndrome, in which there are no familial<br />
cases, additional patients should be studied in order to identify rare<br />
cases with submicroscopic abnormalities, and to pursue a positional<br />
candidate gene approach.<br />
P0291. The role of chromosomal abnormalities in primary<br />
amenorrhoea<br />
B. O. Petrovic, A. Ljubic;<br />
Institute for gynecology and obstetrics, Belgrade, Serbia.<br />
Amenorrhoea is absence or cessation of menses. If menstruation does<br />
not begin by the age of <strong>16</strong> years in the presence of female secondary<br />
sexual maturation, or does not begin by 14 years in the absence<br />
of secondary sexual maturation, the condition is classified as primary<br />
amenorrhoea. This paper deals with investigation of how primary<br />
amenorrhoea and chromosomal abnormalities are related. For the<br />
period of last ten years we have analyzed 62 patients with primary<br />
amenorrhoea. Karyotype from the peripheral blood lymphocytes and<br />
G banding were performed according to standard protocols. Chromosomal<br />
abnormalities were detected in <strong>16</strong> cases (25,8%). Male karyotype<br />
(46,XY) was found in seven cases. In three cases monosomy X<br />
(45,X) was found. In two cases isochromosome X (46,XiXq) was detected,<br />
as well as a case of X chromosome trisomy (47,XXX). Mosaic<br />
karyotypes 45,X/46,XiXq, 46,XX/47,XXY and 45,X/46,XX were found<br />
in one case each. These findings suggest that a consistent number of<br />
cases with primary amenorrhoea (about 25%) can be associated with<br />
genetic anomalies.<br />
P0292. Array-CGH analysis: new deletion syndromes and<br />
atypical phenotype in old deletion syndromes<br />
R. Caselli, F. Mari, F. T. Papa, M. A. Mencarelli, V. Uliana, F. Ariani, I. Meloni, I.<br />
Longo, A. Renieri;<br />
Medical <strong>Genetics</strong>, University of Siena, Siena, Italy.<br />
A group of 28 unrelated MCA/MR patients with normal karyotype, selected<br />
from those attending the Medical <strong>Genetics</strong> Unit of the University<br />
of Siena has been analyzed by oligo array-CGH with an average<br />
resolution of 75Kb (Agilent <strong>Human</strong> Genome CGH Microarray Kit 44B).<br />
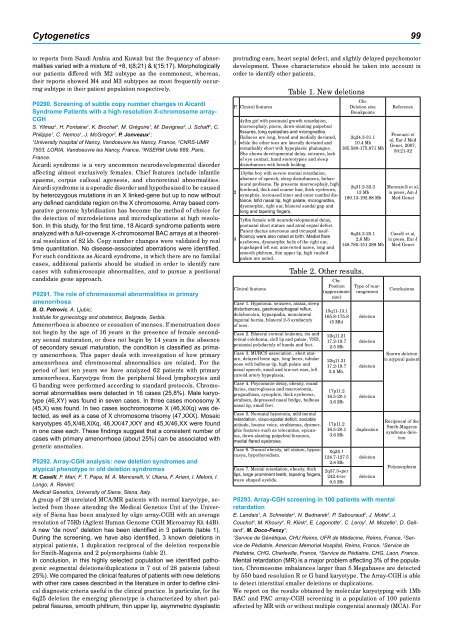
A new “de novo” deletion has been identified in 3 patients (table 1).<br />
During the screening, we have also identified, 3 known deletions in<br />
atypical patients, 1 duplication reciprocal of the deletion responsible<br />
for Smith-Magenis and 2 polymorphisms (table 2).<br />
In conclusion, in this highly selected population we identified pathogenic<br />
segmental deletions/duplications in 7 out of 28 patients (about<br />
25%). We compared the clinical features of patients with new deletions<br />
with other rare cases described in the literature in order to define clinical<br />
diagnostic criteria useful in the clinical practice. In particular, for the<br />
6q25 deletion the emerging phenotype is characterized by short palpebral<br />
fissures, smooth philtrum, thin upper lip, asymmetric dysplastic<br />
protruding ears, heart septal defect, and slightly delayed psychomotor<br />
development. These characteristics should be taken into account in<br />
order to identify other patients.<br />
P. Clinical features<br />
1<br />
2<br />
3<br />
Table 1. New deletions<br />
4y2m girl with postnatal growth retardation,<br />
microcephaly, ptosis, down-slanting palpebral<br />
fissures, long eyelashes and micrognathia.<br />
Halluces are long, broad and medially deviated,<br />
while the other toes are laterally deviated and<br />
remarkably short with hypoplastic phalanges.<br />
She shows developmental delay, seizures, lack<br />
of eye contact, hand stereotypies and sleep<br />
disturbances with breath holding<br />
13y8m boy with severe mental retardation,<br />
absence of speech, sleep disturbances, behavioural<br />
problems. He presents macrocephaly, high<br />
forehead, thick and coarse hair, thick eyebrows,<br />
synophris, increased inner and outer canthal distance,<br />
bifid nasal tip, high palate, micrognathia,<br />
dysmorphic, right ear, bilateral sandal gap and<br />
long and tapering fingers.<br />
7y6m female with neurodevelopmental delay,<br />
postnatal short stature and atrial septal defect.<br />
Patent ductus arteriosus and tricuspid insufficiency<br />
were also noted at birth. Medial flare<br />
eyebrows, dysmorphic helix of the right ear,<br />
cupshaped left ear, anteverted nares, long and<br />
smooth philtrum, thin upper lip, high vaulted<br />
palate are noted.<br />
Clinical features<br />
Case 1. Hypotonia, seizures, ataxia, sleep<br />
disturbances, gastroesophageal reflux,<br />
dolichocolon, hypospadia, monolateral<br />
inguinal hernia, bilateral 2-3 syndactyly<br />
of toes.<br />
Case 2. Bilateral corneal leukoma, iris and<br />
retinal coloboma, cleft lip and palate, VSD,<br />
postaxial polydactyly of hands and feet.<br />
Case 3. MURCS association , short stature,<br />
delayed bone age, long faces, tubular<br />
nose with bulbous tip, high palate and<br />
nasal speech, small and low-set ears, left<br />
carotid artery hypoplasia.<br />
Case 4. Psycomotor delay, obesity, round<br />
facies, macroglossia and macrostomia,<br />
prognathism, synophris, thick eyebrows,<br />
strabism, depressed nasal bridge, bulbous<br />
nasal tip, small feet.<br />
Case 5. Neonatal hypotonia, mild mental<br />
retardation, visuo-spatial deficit, sociable<br />
attitude, hoarse voice, strabismus, dysmorphic<br />
features such as telecantus, epicantus,<br />
down-slanting palpebral fessures,<br />
medial flared eyebrows.<br />
Case 6. Truncal obesity, tall stature, hypoacusya,<br />
hypothyroidism.<br />
Chr.<br />
Deletion size<br />
Breakpoints<br />
2q24.3-31.1<br />
10.4 Mb<br />
<strong>16</strong>5.580-175.871 Mb<br />
2q31.2-32.3<br />
13 Mb<br />
180.13-<strong>19</strong>2.88 Mb<br />
6q24.3-25.1<br />
2,6 Mb<br />
148.785-151.289 Mb<br />
Table 2. Other results.<br />
Chr.<br />
Position<br />
(approximate<br />
size)<br />
15q11-13.1<br />
<strong>16</strong>5.8-175.8<br />
(5 Mb)<br />
22q11.21<br />
17.2-<strong>19</strong>.7<br />
2.5 Mb<br />
22q11.21<br />
17.2-<strong>19</strong>.7<br />
2.5 Mb.<br />
17p11.2<br />
<strong>16</strong>.5-20.1<br />
3.6 Mb<br />
17p11.2<br />
<strong>16</strong>.5-20.1<br />
3.6 Mb<br />
Xq25.1<br />
124.7-127.5<br />
2.8 Mb<br />
Case 7. Mental retardation, obesity, thick<br />
2q37.3-qter<br />
lips, large prominent teeth, tapering fingers,<br />
242.4-ter<br />
wave shaped eyelids.<br />
0.5 Mb<br />
Type of rearrangement<br />
deletion<br />
deletion<br />
deletion<br />
deletion<br />
duplication<br />
deletion<br />
deletion<br />
Reference<br />
Pescucci et<br />
al, Eur J Med<br />
Genet, <strong>2007</strong>,<br />
50:21-32<br />
Mencarelli et al,<br />
in press, Am J<br />
Med Genet<br />
Caselli et al,<br />
in press, Eur J<br />
Med Genet<br />
Conclusions<br />
Known deletion<br />
in atypical patient<br />
Reciprocal of the<br />
Smith-Magenis<br />
syndrome deletion<br />
Polymorphism<br />
P0293. Array-CGH screening in 100 patients with mental<br />
retardation<br />
E. Landais 1 , A. Schneider 1 , N. Bednarek 2 , P. Sabouraud 2 , J. Motte 2 , J.<br />
Couchot 3 , M. Khoury 4 , R. Klink 4 , E. Lagonotte 1 , C. Leroy 1 , M. Mozelle 1 , D. Gaillard<br />
1 , M. Doco-Fenzy 1 ;<br />
1 Service de Génétique, CHU Reims, UFR de Médecine, Reims, France, 2 Service<br />
de Pédiatrie, American Memorial Hospital, Reims, France, 3 Service de<br />
Pédiatrie, CHG, Charleville, France, 4 Service de Pédiatrie, CHG, Laon, France.<br />
Mental retardation (MR) is a major problem affecting 3% of the population.<br />
Chromosome imbalances larger than 5 Megabases are detected<br />
by 550 band resolution R or G band karyotype. The Array-CGH is able<br />
to detect interstitial smaller deletions or duplications.<br />
We report on the results obtained by molecular karyotyping with 1Mb<br />
BAC and PAC array-CGH screening in a population of 100 patients<br />
affected by MR with or without multiple congenital anomaly (MCA). For