

European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Clinical genetics<br />
ing loss (HHL). In many populations mutations in this gene have been<br />
reported as a second cause of HHL. We investigated the prevalence of<br />
SLC26A4 mutations in our HHL consanguineous families. After completing<br />
clinical investigation the signed consent form was taken from<br />
each family. We included 80 families with two or more affected individuals,<br />
who have been referred to GRC. All families who had previously<br />
been tested negative for the DFNB1 locus, were considered as candidates<br />
for homozygosity mapping using STR (Short tandem repeat)s<br />
linked to DFNB4 locus. Families localized to this region were subjected<br />
to complete DNA sequencing for SLC26A4 gene. Ten out of eighty<br />
families were mapped to DFNB4. Sequence analysis of ten linked<br />
families revealed eight mutations in seven families (T420I, 1<strong>19</strong>7delT,<br />
G334Y, R409H, T721M, R79X, S448L, L445W) and<br />
The T420I , G334V and R79X were novel mutations. We have been<br />
able to localize total of 10 families (12.5%) from non-DFNB1 families<br />
to the DFNB4 locus. We detected in all ten families some degrees of<br />
diffuse or nodular goiter, eight out of 10 families showed normal thyroid<br />
function and in six of ten families we found positive prechlorate discharge<br />
test. All of affected had normal temporal bone scan.<br />
This investigation, demonstrated that the SLC26A4 gene mutation is<br />
the most prevalent syndromic hereditary hearing loss in Iran.This result<br />
is in accordance with reports from other countries.<br />
Key words:, SLC26A4, hearing loss,pendred<br />
P0245. Molecular analysis of SMN1 gene common deletions in<br />
some Iranian Spinal muscular Atrophy patients<br />
M. T. Akbari 1,2 , s. zare 1,3 , f. mahjoubi 1,4 ;<br />
1 Akbari Medical Genetic labaratory, No.98, Taleghani Street, Tehran, Islamic<br />
Republic of Iran, 2 Department of Medical <strong>Genetics</strong>,Tarbiat Modares<br />
University,Al-Ahmad Expressway, Tehran, Islamic Republic of Iran, 3 Islamic<br />
Azad Unversity Science and Research Campus, Tehran, Islamic Republic of<br />
Iran, 4 National Research Center for Genetic Engineering & Biotechnology, Tehran,<br />
Islamic Republic of Iran.<br />
Spinal Muscular Atrophy (SMA) is an autosomal recessive neuromuscular<br />
disorder caused by mutations in the SMN1 gene, mainly intragenic<br />
deletions, the commonest of which are deletion in exons 7 and<br />
8. The disorder is subdivided into three clinical groups (type I - III).<br />
The molecular basis for variation in clinical manifestation depends on<br />
the copy number of SMN2 gene in each patient. In this study we present<br />
fifteen families who had at least one live affected SMA patient,<br />
selected for molecular characterization. They fulfilled criteria for inclusion<br />
by demonstration of the characteristic clinical features of SMA<br />
phenotype. These clinical diagnoses were corroborated with EMG and<br />
NCV investigations as well as CPK measurement for the majority of<br />
the cases. The patients DNA samples were prepared from whole blood<br />
by standard salting out method. Characterization of deletions in exons<br />
7 and 8 of SMN 1 gene was carried out by utilizing a pair of mismatched<br />
primers which differentiate between SMN1 and SMN2 genes.<br />
We found deletion in seven patients, about 50%. Five patients had<br />
both exons 7 and 8 deleted and two patients had just exon 7 deletion.<br />
Although the present sample size is small, it may be concluded that<br />
the observed deletions has lower frequency compared to <strong>European</strong><br />
populations which is 95%. Therefore other SMN1 gene defects should<br />
be looked in Iranian patients.<br />
P0246. Case study: “A family with several cases of SMA type 1<br />
due to multiple consanguinous marriages in three consecutive<br />
generations, in the pedigree.”<br />
S. Akbaroghli 1,2 , M. Houshmand 3 , T. Majidizadeh 3 ;<br />
1 Deputy for Cultural Affairs and Prevention of Welfare Organization, Tehran,<br />
Islamic Republic of Iran, 2 Dr. Susan Akbaroghli Genetic Counselling Center,<br />
Tehran, Islamic Republic of Iran, 3 Special Medical Center, Tehran, Islamic Republic<br />
of Iran.<br />
The propand is a three months old floppy boy who is the result of third<br />
degree consanguinous marriage (the first sibling).<br />
At birth his mother had hard NVD and the baby was born with cyanosis.<br />
He had apnea and poor feeding for the first days of his life.<br />
He was hospitalized for 12 days.<br />
His grandparents from both sides have third degree relationships.<br />
He has the typical clinical features and characteristics of SMA type<br />
1.His molecular test for SMA type 1was positive and he has deletion of<br />
exons 7 & 8 of SMN1 gene.<br />
The special point in this family is the existence of several cases of SMA<br />
type 1 due to multiple consanguinous marriages in three consecutive<br />
generations, in the pedigree.<br />
P0247. Clinical forms of Smith-Lemli-Opitz syndrome and their<br />
relation to mutations in the DHCR gene _ classification based<br />
on a group of 50 Polish patients<br />
A. Jezela-Stanek 1 , E. Małunowicz 2 , E. Ciara 1 , E. Popowska 1 , M. Piotrowicz 3 , M.<br />
Gajdulewicz 1 , E. Kostyk 4 , K. Spodar 1 , A. Kruczek 4 , A. Pyrkosz 5 , E. Obersztyn 6 ,<br />
D. Wolnik-Brzozowska 7 , M. Krajewska-Walasek 1 ;<br />
1 Dept. Of Medical <strong>Genetics</strong>, The Children’s Memorial Health Institute, Warsaw,<br />
Poland, 2 Dept. of Laboratory Diagnostics, The Children’s Memorial Health Institute,<br />
Warsaw, Poland, 3 Dept. of Medical <strong>Genetics</strong>, Polish Mother’s Memorial<br />
Hospital - Research Institute, Łódź, Poland, 4 Dept. of Medical <strong>Genetics</strong>, University<br />
Children’s Hospital, Kraków, Poland, 5 Department of General, Molecular<br />
Biology and <strong>Genetics</strong>, Medical University of Silesia, Katowice, Poland, 6 Dept.<br />
of Medical <strong>Genetics</strong>, National Institute of Mother and Child, Warsaw, Poland,<br />
7 Dept. of Medical <strong>Genetics</strong>, Medical University, Poznań, Poland.<br />
Smith-Lemli-Opitz syndrome (SLOS) is a metabolic malformation disorder,<br />
caused by impaired activity of 7-dehydrocholesterol reductase,<br />
the last enzyme in cholesterol biosynthesis. Its best known dysmorphic<br />
features include: microcephaly, ptosis, short and up-turned nose, 2/3<br />
toe syndactyly, and external male genital malformations. Nevertheless,<br />
as the spectrum of observed features is very broad, a question<br />
about phenotype-genotype correlations still emerges.<br />
Poland represents a unique distribution of mutations in the DHCR7<br />
gene. Moreover, one of them is thought to be spreading in our country,<br />
hence our effort to delineate the clinical variability of SLOS in relation<br />
to the causative molecular defect.<br />
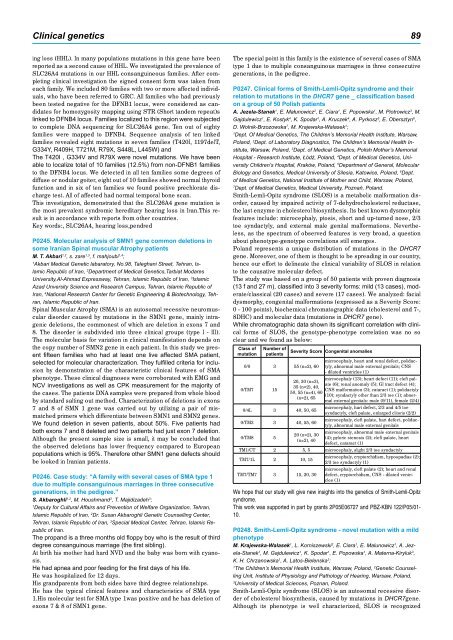
The study was based on a group of 50 patients with proven diagnosis<br />
(13 f and 27 m), classified into 3 severity forms: mild (13 cases), moderate/classical<br />
(20 cases) and severe (17 cases). We analyzed: facial<br />
dysmorphy, congenital malformations (expressed as a Severity Score:<br />
0 - 100 points), biochemical chromatographic data (cholesterol and 7-,<br />
8DHC) and molecular data (mutations in DHCR7 gene).<br />
While chromatographic data shown its significant correlation with clinical<br />
forms of SLOS, the genotype-phenotype correlation was no so<br />
clear and we found as below:<br />
Class of<br />
mutation<br />
Number of<br />
patients<br />
0/0 3 55 (n=2), 60<br />
0/TM7 15<br />
0/4L 3 40, 50, 65<br />
0/TM3 3 40, 55, 60<br />
0/TM8 5<br />
Severity Score Congenital anomalies<br />
microcephaly, heart and renal defect, polidactyly,<br />
abnormal male external genitals; CNS<br />
- dilated ventricles (1)<br />
microcephaly (13); heart defect (11); cleft pal-<br />
20, 30 (n=3),<br />
ate (6); renal anomaly (5); GI tract defect (4);<br />
35 (n=2), 40,<br />
CNS malformation (3); cataract (1); polidactyly<br />
50, 55 (n=4), 60<br />
(10); syndactyly other than 2/3 toe (1); abnor-<br />
(n=2), 65<br />
mal external genitals: male (9/11), female (2/4)<br />
20 (n=2), 30<br />
(n=2), 40<br />
microcephaly, hart defect, 2/3 and 4/5 toe<br />
syndactyly, cleft palate, enlarged clitoris (2/2)<br />
microcephaly, cleft palate, hart defect, polidactyly,<br />
abnormal male external genitals<br />
microcephaly, abnormal male external genitals<br />
(4); pyloric stenosis (2); cleft palate, heart<br />
defect, cataract (1)<br />
TM1/CT 2 5, 5 microcephaly, slight 2/3 toe syndactyly<br />
TM7/1L 2 10, 15<br />
TM7/TM7 3 15, 20, 30<br />
microcephaly, cryptorchidism, hypospadias (2);<br />
2/3 toe syndactyly (1)<br />
microcephaly, cleft palate (2); heart and renal<br />
defect, cryptorchidism, CNS - dilated ventricles<br />
(1)<br />
We hope that our study will give new insights into the genetics of Smith-Lemli-Opitz<br />
syndrome.<br />
This work was supported in part by grants 2P05E06727 and PBZ-KBN 122/P05/01-<br />
10.<br />
P0248. Smith-Lemli-Opitz syndrome - novel mutation with a mild<br />
phenotype<br />
M. Krajewska-Walasek 1 , L. Korniszewski 2 , E. Ciara 1 , E. Malunowicz 1 , A. Jezela-Stanek<br />
1 , M. Gajdulewicz 1 , K. Spodar 1 , E. Popowska 1 , A. Materna-Kiryluk 3 ,<br />
K. H. Chrzanowska 1 , A. Latos-Bielenska 3 ;<br />
1 The Children’s Memorial Health Institute, Warsaw, Poland, 2 Genetic Counselling<br />
Unit, Institute of Physiology and Pathology of Hearing, Warsaw, Poland,<br />
3 University of Medical Sciences, Poznan, Poland.<br />
Smith-Lemli-Opitz syndrome (SLOS) is an autosomal recessive disorder<br />
of cholesterol biosynthesis, caused by mutations in DHCR7gene.<br />
Although its phenotype is well characterized, SLOS is recognized