

European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
Clinical genetics<br />
this form of CDG type II the gene defect has not been discovered yet.<br />
Based on the recently defined clinical syndromes with N- and O-linked<br />
glycosylation defects one should consider the diagnosis of Congenital<br />
Disorders of Glycosylation in patients with a broad spectrum of different<br />
features, including pachygyria, hypotonia with adducted thumbs,<br />
cutis laxa, cardiac and cranio-skeletal anomalies.<br />
P0046. Neurological manifestations of the Cardio-faciocutaneous<br />
Syndrome (CFC)<br />
G. Yoon1 , J. Rosenberg1 , S. Blaser1 , K. Rauen2 ;<br />
1 2 University of Toronto, Toronto, ON, Canada, University of California San Francisco,<br />
San Francisco, CA, United States.<br />
The cardio-facio-cutaneous syndrome (CFC) is a multiple congenital<br />
anomaly disorder characterized by craniofacial dysmorphia, ectodermal<br />
abnormalities, congenital heart defects, developmental delay and<br />
growth retardation. Neurological complications associated with CFC<br />
remain to be clearly defined. Recent discovery of causative mutations<br />
in the MAPK pathway now permit accurate molecular diagnosis of<br />
CFC. Objective: To characterize the neurological features of patients<br />
with molecularly-confirmed CFC. Methods: Medical records, laboratory<br />
and imaging data were reviewed for 37 mutation-positive CFC<br />
patients. Patients with a clinical diagnosis of CFC but a negative result<br />
on mutation screening of the BRAF, KRAS, MEK1 and MEK2 genes<br />
were excluded from the study. Results: Hypotonia, motor delay, speech<br />
delay and learning disability were universally present in this cohort.<br />
Macrocephaly was present in 59%, ptosis in 50%, strabismus in 64%<br />
and nystagmus in 50% of patients. Corticospinal tract findings were<br />
present in 32% of the group. Ventriculomegaly or hydrocephalus was<br />
present in 66% of patients. Other findings on MRI included prominent<br />
Virchow-Robin spaces (<strong>19</strong>%) and abnormal myelination (13%). Seizures<br />
were present in 46% of patients. No specific genotype-phenotype<br />
correlations were observed. Interpretation: Alteration of function<br />
through the Ras/MAPK cascade has traditionally been associated with<br />
oncogenesis. Germline mutations in this pathway have an adverse impact<br />
on neurodevelopment, and appear to play an important role in<br />
ocular function, structural brain anatomy and electrical activity.<br />
P0047. BRAF gene mutation in a patient with cardio-faciocutaneous<br />
(CFC) syndrome presenting with lipoma of corpus<br />
callosum, hepatic and renal cysts<br />
E. Papadopoulou1 , S. Sifakis1 , K. Gripp2 , S. Kirwin2 , K. Sol-Church2 , D. Stabley2<br />
, P. Vorgia1 , M. Kalmanti1 ;<br />
1 2 University Hospital of Heraklion, Heraklion, Greece, Department of Biomedical<br />
Research, Nemours’ Children Clinic, Willmington, DE, United States.<br />
Introduction: Cardio-facio-cutaneous syndrome (CFC) is characterized<br />
by craniofacial features, cardiac defects, ectodermal abnormalities,<br />
and psychomotor delay. It has phenotypic similarities with Noonan and<br />
Costello syndromes.<br />
Case presentation: a 20-month-old boy, the first child of phenotypically<br />
normal parents, who during infancy presented poor suck, and failure to<br />
thrive. At 20th month of life he presents dysmorphic features, postnatal<br />
onset growth deficiency, hypertrophic cardiomyopathy, hypotonia, and<br />
mental retardation. Dysmorphic features include relatively large head,<br />
downslanting palpebral fissures, broad nasal base, protruding nostrils,<br />
high arched palate, papilloma-lile lesion on midline of the fontal region,<br />
deep palmar and plantar creases, sparse curly hair, and hyperextensible<br />
fingers. Brain MRI reveals a lipoma of corpus callosum. Abdomen<br />
ultrasound shows hepatic and renal cysts. DNA analysis initially performed<br />
excluded Noonan and Costello syndromes. Further examination<br />
revealed a 770A-G transition in exon 6 of the BRAF gene, predicting<br />
a gln257-to-arg (Q257R) amino acid change.<br />
Conclusion: CFC syndrome can be caused by gain of function mutations<br />
in 1 of 4 different genes: KRAS, BRAF, MEK1, and MEK2. The<br />
protein products of these genes interact in a common RAS/ERK pathway<br />
that regulates cell differentiation, proliferation, and apoptosis. The<br />
mutation found in our patient has been earlier described in 3 unrelated<br />
CFC patients (BRAF, gln257arg), by Niihori et al. (Nat Genet, 2006).<br />
Hepatomegaly and absence or hypoplasia of corpus callosum has<br />
been described in CFC syndrome. To our knowledge, our patient is the<br />
first who presents lipoma of corpus callosum, as well as hepatic and<br />
renal cystic formations.<br />
P0048. Brain imaging in patients with KRAS, BRAF and MEK<br />
mutations<br />
C. Nava 1 , C. Michot 1 , N. Hanna 2 , S. Pereira 1 , B. Parfait 2 , J. Elion 1 , A. Verloes 1 ,<br />
H. Cavé 1 ;<br />
1 department of genetics APHP hopital Robert Debré, Paris, France, 2 Inserm<br />
U745, Faculté des Sciences Pharmaceutiques et Biologiques, Université Paris<br />
V, Paris, France.<br />
Cardio-facio-cutaneous syndrome (CFC) is a severe developmental<br />
disorder clinically related to Noonan syndrome and Costello syndrome<br />
(CS), which has been recently linked to mutations in 4 genes of the<br />
RAS/MEK/ERK signaling pathway. Developmental delay is frequent in<br />
those diseases but abnormalities of brain imaging aren’t often reported<br />
in these patients. In this study, we performed mutation analysis of<br />
KRAS, BRAF, MEK1, and MEK2 in 40 patients with a CFC, 20 patients<br />
with a CS without HRAS mutations, and 82 patients with NS without<br />
PTPN11 mutations. We found 22 BRAF mutations, 7 KRAS mutations,<br />
11 MEK1 mutations, and 5 MEK2 mutations. 21/45 patients with a mutation<br />
had a brain imagery. It was abnormal in 11/21 of cases (table 1).<br />
5/14 CS with a mutation in KRAS, BRAF or MEK had abnormal MRI (4<br />
patients: normal, 5 patients : unrealized). Cerebral malformations were<br />
less frequent in CFC patients (abnormalities in 3/23, 5 normal, 15 unrealized).<br />
3 patients with NS present a ventricular dilatation associated<br />
with a cerebral atrophy.<br />
Brain abnormalities are frequent in all gene mutated and clinical diagnosis<br />
and a brain imaging seems to be justified in medical care of<br />
these patients.<br />
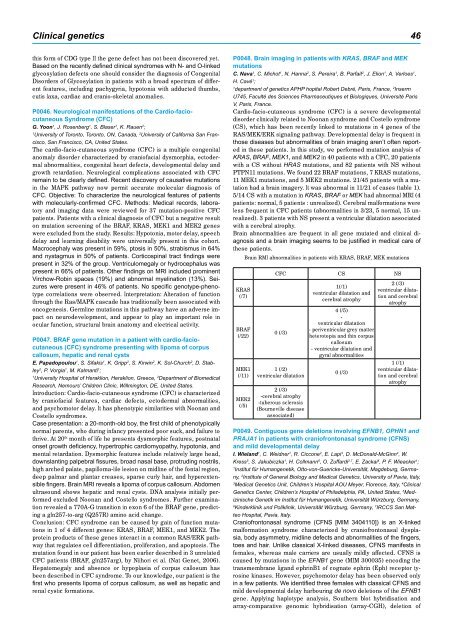
KRAS<br />
(/7)<br />
BRAF<br />
(/22)<br />
MEK1<br />
(/11)<br />
MEK2<br />
(/5)<br />
Brain RMI abnormalities in patients with KRAS, BRAF, MEK mutations<br />
CFC CS NS<br />
0 (/3)<br />
1 (/2)<br />
ventricular dilatation<br />
2 (/3)<br />
-cerebral atrophy<br />
-tuberous sclerosis<br />
(Bourneville disease<br />
associated)<br />
1(/1)<br />
ventricular dilatation and<br />
cerebral atrophy<br />
4 (/5)<br />
-<br />
ventricular dilatation<br />
- periventricular grey matter<br />
heterotopia and thin corpus<br />
callosum<br />
- ventricular dilatation and<br />
gyral abnormalities<br />
0 (/3)<br />
2 (/3)<br />
ventricular dilatation<br />
and cerebral<br />
atrophy<br />
1 (/1)<br />
ventricular dilatation<br />
and cerebral<br />
atrophy<br />
P0049. Contiguous gene deletions involving EFNB1, OPHN1 and<br />
PRAJA1 in patients with craniofrontonasal syndrome (CFNS)<br />
and mild developmental delay<br />
I. Wieland1 , C. Weidner1 , R. Ciccone2 , E. Lapi3 , D. McDonald-McGinn4 , W.<br />
Kress5 , S. Jakubiczka1 , H. Collmann6 , O. Zuffardi2,7 , E. Zackai4 , P. F. Wieacker1 ;<br />
1Institut für <strong>Human</strong>genetik, Otto-von-Guericke-Universität, Magdeburg, Germany,<br />
2Institute of General Biology and Medical <strong>Genetics</strong>, University of Pavia, Italy,<br />
3 4 Medical <strong>Genetics</strong> Unit, Children’s Hospital AOU Meyer, Florence, Italy, Clinical<br />
<strong>Genetics</strong> Center, Children’s Hospital of Philadelphia, PA, United States, 5Med izinische Genetik im Institut für <strong>Human</strong>genetik, Universität Würzburg, Germany,<br />
6 7 Kinderklinik und Poliklinik, Universität Würzburg, Germany, IRCCS San Matteo<br />
Hospital, Pavia, Italy.<br />
Craniofrontonasal syndrome (CFNS [MIM 3404110]) is an X-linked<br />
malformation syndrome characterized by craniofrontonasal dysplasia,<br />
body asymmetry, midline defects and abnormalities of the fingers,<br />
toes and hair. Unlike classical X-linked diseases, CFNS manifests in<br />
females, whereas male carriers are usually mildly affected. CFNS is<br />
caused by mutations in the EFNB1 gene (MIM 300035) encoding the<br />
transmembrane ligand ephrinB1 of cognate ephrin (Eph) receptor tyrosine<br />
kinases. However, psychomotor delay has been observed only<br />
in a few patients. We identified three females with classical CFNS and<br />
mild developmental delay harbouring de novo deletions of the EFNB1<br />
gene. Applying haplotype analysis, Southern blot hybridisation and<br />
array-comparative genomic hybridisation (array-CGH), deletion of