

European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...
European Human Genetics Conference 2007 June 16 – 19, 2007 ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Molecular and biochemical basis of disease<br />
P0774. IRF gene’s nucleotide sequence changes in patients<br />
with nonsyndromic orofacial clefting from Lithuania<br />
A. Morkūnienė 1,2 , L. Ambrozaitytė 1 , A. Utkus 1,2 , V. Kučinskas 1,2 ;<br />
1 Vilnius University, Faculty of Medicine, Department of <strong>Human</strong> and Medical<br />
<strong>Genetics</strong>, Vilnius, Lithuania, 2 Centre for Medical <strong>Genetics</strong>, Vilnius University<br />
Hospital Santariškių Klinikos, Vilnius, Lithuania.<br />
The hunt for the causes of nonsyndromic orofacial clefing (NS-OFC)<br />
is extremely complex, involving multiple techniques that have been<br />
used to identify numerous candidate genes in which disruption results<br />
in increased risk of clefting. In recent years, a number of independent<br />
groups have targeted for investigation the involvement of IRF6 (interferon<br />
regulatory factor 6 gene). IRF6 is related to syndromic OFC - Van<br />
der Woude syndrome.<br />
Our study was aimed to investigate whether mutations in the IRF6<br />
gene contribute to NS-OFC in the population of Lithuania.<br />
Patients with NS-OFCs from Lithuania were tested for nucleotide sequence<br />
changes in the IRF6 gene (206 patients). DNA fragments covering<br />
exonic parts of the IRF6 gene were PCR-amplified and direct<br />
sequenced.<br />
23 different nucleotide sequence changes were revealed in the IRF6<br />
gene by comparison of sequencing results with reference DNA sequences<br />
of the genes. Scanning IRF6 gene resulted in ten novel<br />
nucleotide sequence variants. Out of them, four were missense mutations<br />
(p.S212I, p.L295P, p. Q340K, p.R400L), which, together with the<br />
p.A61G mutation found in the case of Van der Woude syndrome, might<br />
be related to the NS-OFC phenotype in the population of Lithuania.<br />
Our study highlights the IRF6 gene sequence variability and supports<br />
the hypothesis that variation in this gene contributes to NS-OFC phenotype<br />
encouraging further investigations to test if IRF6 gene mutations<br />
identified in the individuals from Lithuania are rare alleles causative<br />
for NS-OFC.<br />
P0775. The R176C amino-acid change in hemojuvelin as a<br />
novel haemochromatosis mutation: phenotypic and functionanl<br />
evidences<br />
C. Ka 1 , G. Le Gac 1 , E. Letocart 1 , I. Gourlaouen 1 , L. Bryckaert 1 , B. Martin 2 , C.<br />
Ferec 1 ;<br />
1 Inserm, U613; Univ Bretagne Occidentale; Etablissement Français du Sang,<br />
Brest, F-29200, France, 2 Etablissement Français du Sang, Niort, F-79000,<br />
France.<br />
Background: Juvenile haemochromatosis (JH) is an early-onset autosomal<br />
recessive condition of iron metabolism caused by mutations in<br />
either the hemojuvelin (HJV) or the hepcidin-encoding gene (HAMP).<br />
The two JH gene products are implicated in a same physiologic process,<br />
where hepcidin controls iron flow into plasma and hemojuvelin<br />
enhances hepcidin expression at the transcriptional level via the classical<br />
bone morphogenetic protein (BMP) cell signalling pathway. Aim<br />
of the study and results: In this study, we report a novel missense<br />
HJV mutation that leads to the replacement of arginine by a cysteine<br />
residue at position 176 (R176C). We associate homozygosity for this<br />
novel mutation with the iron overload phenotype observed in a 17year-old<br />
girl. We also show that the HJV 176C mutated protein fails<br />
to up-regulate the hepcidin promoter activity. Lastly, we suggest that,<br />
due to its nature and position, the R176C amino-acid change prevents<br />
an autocatalytic cleavage that normally occurs during HJV intracellular<br />
processing. Conclusion: Our results definitively demonstrate that the<br />
R176C substitution is a novel HJV loss-of-function mutation. They also<br />
highlight that rapid advances in comprehension of the HJV intracellular<br />
processing and function have paved the way for functional characterizations.<br />
P0776. Early age at onset is the major clinical feature in<br />
Parkinson disease related to PARK2 gene mutations<br />
C. Cazeneuve 1 , C. San 1 , E. Lohmann 2 , S. Lesage 2 , E. Leguern 1,2 , A. Durr 3,2 , A.<br />
Brice 4,2 , the Parkinson Disease <strong>Genetics</strong> Group;<br />
1 Dpt of <strong>Genetics</strong>, Neurogenetics laboratory, Hospital Pitié-Salpêtrière, Paris,<br />
France, 2 Institut National de la Santé et de la Recherche Médicale Unit U679,<br />
Hospital Pitié-Salpêtrière, Paris, France, 3 Dpt of <strong>Genetics</strong>, Clinical <strong>Genetics</strong>,<br />
Paris, France, 4 Dpt of <strong>Genetics</strong>, Hospital Pitié-Salpêtrière, Paris, France.<br />
Autosomal recessive juvenile Parkinson disease (JPD) is characterized<br />
by rigidity, bradykinesia, rest tremor, age at onset before 40 years,<br />
good response to levodopa treatment, and slowly progressive disease.<br />
Mutations in the parkin gene (PARK2) are the major genetic cause of<br />
20<br />
JPD.<br />
To refine the clinical characteristics of the PARK2-associated JPD,<br />
we investigated 77 patients referred to our diagnostic lab for PARK2<br />
analysis.<br />
Searching for point mutations by sequencing and for large rearrangements<br />
by Multiplex Ligation-dependent Probe Amplification (MLPA kits<br />
P051/P052, MRC-Holland) gave the following results:<br />
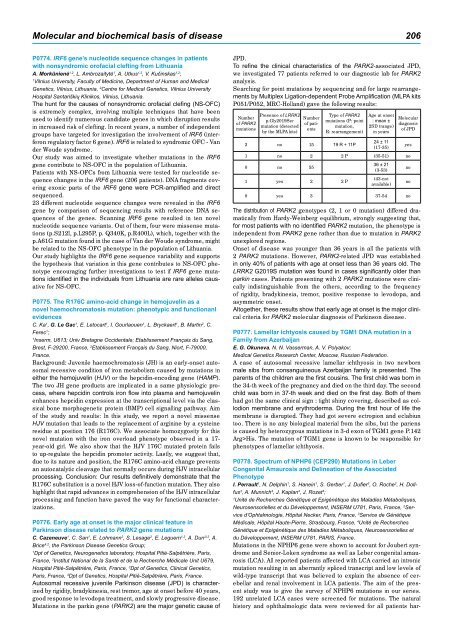
Number<br />
of PARK2<br />
mutations<br />
Presence of LRRK2<br />
p.Gly20<strong>19</strong>Ser<br />
mutation (detected<br />
by the MLPA kits)<br />
Number<br />
of patients<br />
Type of PARK2<br />
mutations (P: point<br />
mutation,<br />
R: rearrangement)<br />
2 no 15 <strong>19</strong> R + 11P<br />
Age at onset<br />
: mean ±<br />
2SD (range)<br />
in years<br />
24 ± 11<br />
(17-35)<br />
Molecular<br />
diagnosis<br />
of JPD<br />
1 no 2 2 P (35-51) no<br />
0 no 55<br />
1 yes 2 2 P<br />
36 ± 21<br />
(3-53)<br />
(43-not<br />
available)<br />
0 yes 3 37-54 no<br />
The distribution of PARK2 genotypes (2, 1 or 0 mutation) differed dramatically<br />
from Hardy-Weinberg equilibrium, strongly suggesting that,<br />
for most patients with no identified PARK2 mutation, the phenotype is<br />
independent from PARK2 gene rather than due to mutation in PARK2<br />
unexplored regions.<br />
Onset of disease was younger than 36 years in all the patients with<br />
2 PARK2 mutations. However, PARK2-related JPD was established<br />
in only 40% of patients with age at onset less than 36 years old. The<br />
LRRK2 G20<strong>19</strong>S mutation was found in cases significantly older than<br />
parkin cases. Patients presenting with 2 PARK2 mutations were clinically<br />
indistinguishable from the others, according to the frequency<br />
of rigidity, bradykinesia, tremor, positive response to levodopa, and<br />
asymmetric onset.<br />
Altogether, these results show that early age at onset is the major clinical<br />
criteria for PARK2 molecular diagnosis of Parkinson disease.<br />
P0777. Lamellar ichtyosis caused by TGM1 DNA mutation in a<br />
Family from Azerbaijan<br />
E. G. Okuneva, N. N. Vasserman, A. V. Polyakov;<br />
Medical <strong>Genetics</strong> Research Center, Moscow, Russian Federation.<br />
A case of autosomal recessive lamellar ichthyosis in two newborn<br />
male sibs from consanguineous Azerbaijan family is presented. The<br />
parents of the children are the first cousins. The first child was born in<br />
the 34-th week of the pregnancy and died on the third day. The second<br />
child was born in 37-th week and died on the first day. Both of them<br />
had got the same clinical sign : tight shiny covering, described as collodion<br />
membrane and erythroderma. During the first hour of life the<br />
membrane is disrupted. They had got severe ectropion and eclabiun<br />
too. There is no any biological material from the sibs, but the pariens<br />
is caused by heterozygous mutations in 3-d exon of TGM1 gene P.142<br />
Arg>His. The mutation of TGM1 gene is known to be responsible for<br />
phenotypes of lamellar ichthyosis.<br />
P0778. Spectrum of NPHP6 (CEP290) Mutations in Leber<br />
Congenital Amaurosis and Delineation of the Associated<br />
Phenotype<br />
I. Perrault 1 , N. Delphin 1 , S. Hanein 1 , S. Gerber 1 , J. Dufier 2 , O. Roche 2 , H. Dollfus<br />
3 , A. Munnich 4 , J. Kaplan 4 , J. Rozet 4 ;<br />
1 Unité de Recherches Génétique et Epigénétique des Maladies Métaboliques,<br />
Neurosensorielles et du Développement, INSERM U781, Paris, France, 2 Service<br />
d’Ophtalmologie, Hôpital Necker, Paris, France, 3 Service de Génétique<br />
Médicale, Hôpital Haute-Pierre, Strasbourg, France, 4 Unité de Recherches<br />
Génétique et Epigénétique des Maladies Métaboliques, Neurosensorielles et<br />
du Développement, INSERM U781, PARIS, France.<br />
Mutations in the NPHP6 gene were shown to account for Joubert syndrome<br />
and Senior-Loken syndrome as well as Leber congenital amaurosis<br />
(LCA). All reported patients affected with LCA carried an intronic<br />
mutation resulting in an aberrantly spliced transcript and low levels of<br />
wild-type transcript that was believed to explain the absence of cerebellar<br />
and renal involvement in LCA patients. The aim of the present<br />
study was to give the survey of NPHP6 mutations in our series.<br />
<strong>19</strong>2 unrelated LCA cases were screened for mutations. The natural<br />
history and ophthalmologic data were reviewed for all patients har-<br />
yes<br />
no<br />
no