

ASE Manual Release 3.6.1.2825 CAMd - CampOS Wiki
ASE Manual Release 3.6.1.2825 CAMd - CampOS Wiki
ASE Manual Release 3.6.1.2825 CAMd - CampOS Wiki

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
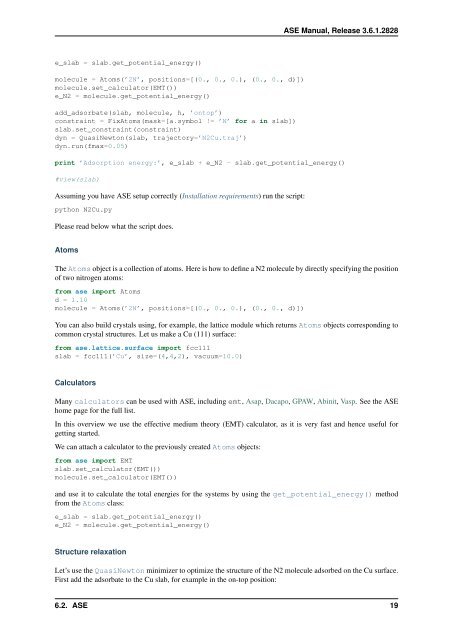
e_slab = slab.get_potential_energy()<br />
molecule = Atoms(’2N’, positions=[(0., 0., 0.), (0., 0., d)])<br />
molecule.set_calculator(EMT())<br />
e_N2 = molecule.get_potential_energy()<br />
add_adsorbate(slab, molecule, h, ’ontop’)<br />
constraint = FixAtoms(mask=[a.symbol != ’N’ for a in slab])<br />
slab.set_constraint(constraint)<br />
dyn = QuasiNewton(slab, trajectory=’N2Cu.traj’)<br />
dyn.run(fmax=0.05)<br />
<strong>ASE</strong> <strong>Manual</strong>, <strong>Release</strong> 3.6.1.2828<br />
print ’Adsorption energy:’, e_slab + e_N2 - slab.get_potential_energy()<br />
#view(slab)<br />
Assuming you have <strong>ASE</strong> setup correctly (Installation requirements) run the script:<br />
python N2Cu.py<br />
Please read below what the script does.<br />
Atoms<br />
The Atoms object is a collection of atoms. Here is how to define a N2 molecule by directly specifying the position<br />
of two nitrogen atoms:<br />
from ase import Atoms<br />
d = 1.10<br />
molecule = Atoms(’2N’, positions=[(0., 0., 0.), (0., 0., d)])<br />
You can also build crystals using, for example, the lattice module which returns Atoms objects corresponding to<br />
common crystal structures. Let us make a Cu (111) surface:<br />
from ase.lattice.surface import fcc111<br />
slab = fcc111(’Cu’, size=(4,4,2), vacuum=10.0)<br />
Calculators<br />
Many calculators can be used with <strong>ASE</strong>, including emt, Asap, Dacapo, GPAW, Abinit, Vasp. See the <strong>ASE</strong><br />
home page for the full list.<br />
In this overview we use the effective medium theory (EMT) calculator, as it is very fast and hence useful for<br />
getting started.<br />
We can attach a calculator to the previously created Atoms objects:<br />
from ase import EMT<br />
slab.set_calculator(EMT())<br />
molecule.set_calculator(EMT())<br />
and use it to calculate the total energies for the systems by using the get_potential_energy() method<br />
from the Atoms class:<br />
e_slab = slab.get_potential_energy()<br />
e_N2 = molecule.get_potential_energy()<br />
Structure relaxation<br />
Let’s use the QuasiNewton minimizer to optimize the structure of the N2 molecule adsorbed on the Cu surface.<br />
First add the adsorbate to the Cu slab, for example in the on-top position:<br />
6.2. <strong>ASE</strong> 19









