Diploma - Max Planck Institute for Solid State Research
Diploma - Max Planck Institute for Solid State Research
Diploma - Max Planck Institute for Solid State Research

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
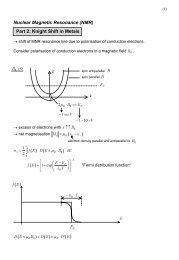
8 2 Theoretical foundation<br />
such a system anyway (<strong>for</strong> example as a zeroth order approximation), one has to challenge<br />
additionally the convergence instability of the Fermi level determination process,<br />
since the dispersion of the localized states is weak and there<strong>for</strong>e a small rearrangement<br />
of the Fermi level changes the occupation number strongly. A possible solution is<br />
to modify the functional so that it contains some correction to the correlation energy<br />
(e.g. L(S)DA+U). To circumvent this issue the localized electrons have been treated as<br />
“core” electrons 1 (so-called open core approximation) neglecting the overlap from different<br />
sites. This approximation is legitimate, because their magnitude of localization<br />
is comparable to orbitals treated as “core” electrons, but their single-particle energy is<br />
considerably higher. Moreover, in ch. 4.1 it is shown, that the Fermi level obtained<br />
by this method is comparable to L(S)DA+U results with respect to the valence band<br />
structure.<br />
2.1.3 Codes<br />
There are a lot of DFT codes available with various approximations depending on the<br />
implemented basis set (a few implementations of the respecting methods are given at<br />
the end of each block). In general, a distinction[17, p. 233–235] can be drawn between<br />
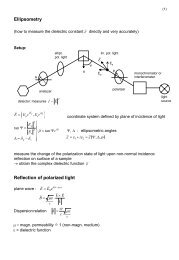
(a) Plane wave methods present the most general way <strong>for</strong> solving differential equations.<br />
It is easy to implement them <strong>for</strong> computation and since being the solution of<br />
the Schrödinger equation with constant potential, they are an effective basis <strong>for</strong> the<br />
nearly-free electron model (covering the crystal potential as a small perturbation)<br />
there<strong>for</strong>e one gets a valuable insight to the bandstructure of sp-metals and semiconductors.<br />
The disadvantage is enclosed in the potential representation because plane<br />
waves demand a smooth potential whereas the Coulomb potential has a singularity.<br />
Hence, those methods are often accompanied by pseudopotentials (smoothed<br />
potentials, nucleus and core electrons are combined) or grids.<br />
(e.g. Abinit [http://www.abinit.org], VASP [http://cms.mpi.univie.ac.at/vasp], Quantum-<br />
Expresso [http://www.quantum-espresso.org], CPMD [http://www.cpmd.org], ...)<br />
(b) Choosing localized (atomic-like) orbitals as a basis set respects automatically<br />
the symmetry in the vicinity of the atomic sites. As basis functions of the Bloch<br />
states are usually selected Gaussians, or numerically adjusted atomic-like orbitals<br />
(demands Bloch Theorem, cf. (2.11), (2.12)). The advantage of being able to use<br />
the bare Coulomb potential (superposition of the atomic Coulomb potentials) is<br />
gaining high accuracy <strong>for</strong> heavier elements as well as having a smaller basis set<br />
compared to plane wave methods. Since atomic orbitals from different sites are not<br />
1 Further on, this approximation will be called “open core approximation”, in literature also frozen core<br />
or quasi-core, because we deal with not fully-occupied “core” electrons. Since it is not a stable noble<br />
gas configuration, the occupation number should in principle be determined variationally. Given<br />
that the overlap of the localized orbitals is small, one can set a fixed occupation number as an initial<br />
parameter according to the experiment