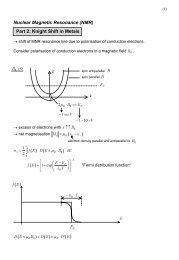
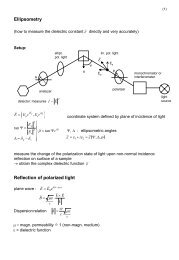
Diploma - Max Planck Institute for Solid State Research
Diploma - Max Planck Institute for Solid State Research
Diploma - Max Planck Institute for Solid State Research

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
4.1 Overview – properties and classification 29<br />
the charge density a first time. Afterwards, the self-consistency cycle of the occupation<br />
matrix has been permitted and the final result obtained. Returning to fig. 4.4, the<br />
characteristica of this iterative process will be discussed. Fixing the occupation matrix<br />
of the correlated orbital avoids shifting the orbital energies during the iterative process<br />
due to the model Hamiltonian. Hence the eigenenergies of all 4f orbitals are located near<br />
the Fermi level (red dotted representation). Allowing the fully self-consistent treatment<br />
shifts the occupied (unoccupied) 4f orbitals below (above) the Fermi level, respectively<br />
(blue dashed representation). Having 7 localized orbitals (spin degenerate) with an<br />
occupation of 6.7, at least 3 should lie below the Fermi level (since their dispersion is<br />
negligible). One can see, that <strong>for</strong> the self-consistent LDA+U calculation one 4f orbital<br />
is 3 eV, one 2 eV below the Fermi level and two are around the Fermi level (being<br />
partially above). Regarding regions marked by 1, it is apparent that the open core<br />
approximation is suitable <strong>for</strong> the VB in an energy range not allocated by 4f orbitals.<br />
In difference comparing 2a to 2b (being in the range of localized states), the symmetry<br />
of the VB determines the hybridization strength, and thus the deviation from the open<br />
core calculation. There<strong>for</strong>e it is necessary to apply a hybridization model (see ch. 4.2)<br />
to rearrange the band structure properly in the open core approximation (e.g. regarding<br />
the Fermi surface of the compound).<br />
4.1.3 Cleavage behaviour<br />
As already mentioned in ch. 3, the surface is prepared in-situ by sample splitting, hence<br />
the structure has several rupture lines (since it is layered). The lever stick has been<br />
oriented in the [001] direction, so that we were able to measure the [001] surface BZ in<br />
both setups (because k x , k y are equivalent and there<strong>for</strong>e the orientation of the entrance<br />
slit does only matter <strong>for</strong> the type of polarization). To estimate the possible cleavage<br />
plane and thus the atom type of the surface layer (termination) two different approaches<br />
have been used:<br />
(a) comparison of bond strength<br />
The bond strength between the layers can be estimated by the “charge transfer”<br />
inside the solid compared to the atomic allocation because electrons mediate bonds<br />
which implies that the iso-charge density is a valid measure <strong>for</strong> their distribution.<br />
There<strong>for</strong>e the sum of the atomic charge densities (<strong>for</strong> each site) has been substracted<br />
from the final (converged) charge density, so positive (negative) remaining charge<br />
density corresponds to an inflow (outflow) of electrons. If one compares Fig. 4.5b<br />
to fig. 4.5c it becomes evident, that the charge density is redistributed from the<br />
Eu layer and the region between Eu and Si to the composition of Si-Rh-Si, mostly<br />
between the atoms Rh-Si / Si-Rh, exactly in bonding direction. Thus the structural<br />
integrity of the latter is larger than that of the Eu-Si bond and one expects either Si<br />
or Eu terminated surfaces. Auxiliary, one can discuss the bonding type evaluating