11 BIOTEC GenMol 10_11 tripl
11 BIOTEC GenMol 10_11 tripl
11 BIOTEC GenMol 10_11 tripl

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Espansione di ripetizioni<br />
Lezione <strong>11</strong><br />
1
Anticipazione<br />
alcuni fenotipi a trasmissione autosomica dominante diventano<br />
piu’ gravi nelle generazioni successive perche?<br />
espansione di <strong>tripl</strong>ette: tipo di mutazione<br />
individuata nel 1990<br />
Corea di Hunghinton, X fragile, Distrofia Miotonica…………<br />
premutazione : stato che aumenta il rischio di trasmettere<br />
il fenotipo patologico. Il rischio non e’ piu’ quantizzabile in<br />
termini mendeliani<br />
prudenza nel considerare neutri i polimorfismi<br />
2
Polimorfismo PCR<br />
Mini e micro satelliti: Riconosciuti su gel dopo amplificazione con<br />
PCR<br />
3
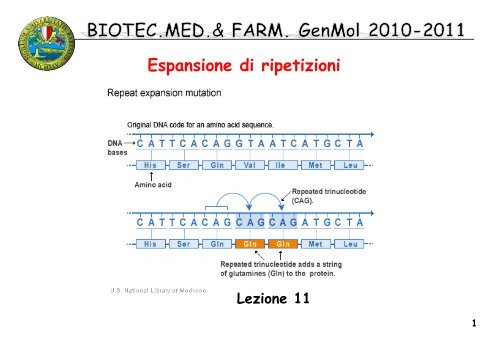
Espansioni di <strong>tripl</strong>ette instabili:<br />
mutazioni dinamiche<br />
Mutazioni dovute all’espansione di trinucleotidi localizzati nelle regioni<br />
codificanti o non-codificanti di alcuni geni, si originano per errori nella<br />
replica del DNA. Possono originare un sito fragile visibile citogeneticamente.<br />
Se si trova in una regione codificante il prodotto del gene<br />
presentera’ n numero di residui amminoacidici corrispondenti<br />
alla <strong>tripl</strong>etta superiori al prodotto dell’allele selvatico. Di solito la <strong>tripl</strong>etta<br />
coinvolta e’ CAG (acido glutammico).<br />
Se si trova in una regione non-codificante possono alterare l’espressione<br />
del gene in cis. Distrofia miotonica: 3’UTR; atassia di Friedrich: introne1;<br />
X-fragile:5’UTR……..<br />
4
Mutazioni dinamiche: origine<br />
Le <strong>tripl</strong>ette possibili sono 64, ma in realta’ le<br />
combinazioni sono solo <strong>10</strong> (per la lettura su entrambi i<br />
filamenti):<br />
AAC/GTT<br />
AAG/CTT<br />
AAT/ATT<br />
ACC/GGT<br />
ACG/CGT<br />
ACT/AGT<br />
AGG/CCT<br />
ATC/GAT<br />
CAG/CTG<br />
CCG/CGG<br />
Tutte le altre sono permutazioni cicliche di queste<br />
5
Mutazioni dinamiche: caratteristiche comuni<br />
La variabilita’ degli alleli ripetuti dipende dalla loro lunghezza<br />
Il tasso di mutazione dipende dalla lunghezza<br />
Le unita’ ripetute tendono ad aumentare piuttosto che a<br />
diminuire<br />
Presenza dello stato di premutazione: espansione di <strong>tripl</strong>ette non<br />
ancora associato alla comparsa del fenotipo, ma che rende la regione<br />
instabile e piu’ suscettibile a raggiungere il livello di mutazione<br />
completa.<br />
Difficolta’ nel definire la correlazione genotipo fenotipo per la<br />
presenza di un range di espansione che si puo’associare alla comparsa<br />
del fenotipo.<br />
Presenza di anticipazione: aumento della gravita’ del fenotipo<br />
nelle generazioni successive<br />
6
Mutazione<br />
completa,<br />
range di<br />
mutazione<br />
Premutazione<br />
Normale<br />
Schema di mutazioni dinamiche<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
200<br />
60<br />
<strong>10</strong>00<br />
280<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
200<br />
GAA<br />
GAA<br />
GAA 22<br />
GAA<br />
GAA<br />
900<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
52<br />
GAA<br />
CTG<br />
GAA<br />
6<br />
7<br />
CTG<br />
GAA CTG<br />
5’UTR esone introne esone introne esone 3’UTR<br />
X-fragile<br />
Atassia di<br />
Friedrich<br />
CTG<br />
37<br />
3<br />
50<br />
Distrofia<br />
miotonica<br />
3000<br />
Mutazione<br />
completa,<br />
range di<br />
mutazione<br />
Premutazione<br />
Normale<br />
CAG<br />
CAG<br />
CAG<br />
CAG 36<br />
CAG<br />
CAG 35<br />
CAG<br />
CAG<br />
CAG<br />
<strong>10</strong><br />
121<br />
CGG<br />
CGG<br />
CGG<br />
CGG 25<br />
Esone ORF<br />
Corea di Huntington Atassia spinocerebellare 1<br />
CGG<br />
CGG<br />
CGG<br />
7<br />
75<br />
49<br />
7
Mutazioni dinamiche: effetti I<br />
Perche’ l’espansione origina un fenotipo patologico?<br />
Espansioni molto ampie esterne alla sequenza codificante:<br />
perdita di funzione.<br />
Espansioni modeste di CAG nelle regioni codificanti<br />
La premutazione e’ uno stato di suscettibilita’: l’instabilita’<br />
puo’ essere considerata una patologia della macchina della<br />
replicazione .<br />
8
Mutazioni dinamiche: effetti II<br />
Perche’ l’espansione origina un fenotipo patologico?<br />
Espansioni molto ampie esterne alla sequenza codificante: perdita di funzione.<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
CGG<br />
200<br />
60<br />
52<br />
6<br />
<strong>10</strong>00<br />
280<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
GAA<br />
7<br />
22<br />
900<br />
200<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
5’UTR esone introne esone introne esone 3’UTR<br />
X-fragile Atassia di Friedrich Distrofia miotonica<br />
CTG<br />
CTG<br />
CTG<br />
CTG<br />
37<br />
3<br />
50<br />
Possono abolire o interferire con la trascrizione.<br />
Sono presenti anche mutazioni puntiformi nella regione codificante che hanno lo stesso<br />
effetto.<br />
Il fenotipo e’ sempre recessivo eccetto per la distrofia miotonica e la SCA8.<br />
A questa classe appartiene il gene per l’epilessia mioclonica giovanile (JME) che e’<br />
originata da espansionedi 12 monomeri (CCCCGCCCCGCG)n<br />
9
Mutazioni dinamiche: effetti III<br />
Perche’ l’espansione origina un fenotipo patologico?<br />
Espansioni modeste di CAG nelle regioni codificanti<br />
CAG<br />
CAG<br />
CAG<br />
CAG<br />
CAG<br />
CAG 35<br />
CAG<br />
CAG<br />
CAG<br />
<strong>10</strong><br />
121<br />
36<br />
CGG<br />
CGG<br />
CGG<br />
CGG 25<br />
Esone ORF<br />
Corea di Huntington Atassia spinocerebellare 1<br />
CGG<br />
CGG<br />
CGG<br />
7<br />
75<br />
49<br />
Codificano tratti di poliglutammina nel prodotto del gene. Si<br />
determina l’aggregazione delle molecole e la morte cellulare.<br />
<strong>10</strong>
Mutazioni dinamiche: effetti IV<br />
Espansioni modeste di CAG nelle regioni codificanti<br />
Caratteri comuni alle malattie di questa classe<br />
Il fenotipo ha trasmissione dominante eccetto per la malattia di Kennedy<br />
(X-linked) e sono patologie neuroderegenitive ad insorgenza tardiva<br />
Malattia di Kennedy o atrofia muscolare spino bulbare. Il gene responsabile<br />
è il recettore degli androgeni<br />
L’allele espanso viene trascritto e la ripetizione codifica per un tratto di<br />
poliglutammina<br />
<strong>11</strong>
X-fragile<br />
Ripeto che questo e’ solo un modello, per spiegare il<br />
fenomeno e capire come funziona il nostro genoma<br />
1/4000 maschi e 1/7000 femmine<br />
È la causa di ritardo mentale più importante nei maschi<br />
12
Metafase di affetto da X-fragile<br />
13
Sindrome X fragile<br />
E’ originata dalla mancata espressione del gene FMR1<br />
Il prodotto del gene lega diversi mRNA ed e’ importante per la<br />
loro corretta traduzione.<br />
Il cromosoma X della quasi totalita’ degli affetti presenta un sito<br />
fragile in Xq28 in una percentuale variabile delle metafasi, se la<br />
coltura di sangue viene eseguita aggiungendo particolari sostanze al<br />
terreno coltura.<br />
Nelle femmine di solito non si riesce ad evidenziare.<br />
Solo una piccola percentuale delle sicure portatrici manifesta il<br />
ritardo mentale<br />
14
X fragile<br />
15
Modalita’ di trasmissione X-fragile<br />
Dal momento che il locus mappa sul<br />
cromosoma X dovrebbe seguire un pattern<br />
di trasmissione X-linked<br />
E’ cosi’, ma analizzando le famiglie e eseguendo<br />
analisi di linkage,prima dell’identificazione del tipo di<br />
mutazione e del clonaggio del gene, emerse quello<br />
che fu chiamato paradosso di Sherman, che porto’ a<br />
ipotizzare la presenza della premutazione<br />
16
Paradosso di Sherman 1985<br />
...... the first mutation caused a “premutation” state<br />
that produced no clinical symptoms, and that a second<br />
mutation was required to convert the premutation to a<br />
“full mutation” form that was associated with the<br />
characteristic symptoms<br />
17
Paradosso di Sherman<br />
La figlia III di un maschio II non affetto, ma portatore, ha un<br />
rischio più elevato di avere figli affetti rispetto a sua nonna I<br />
anche se di fatto ha il suo “stesso” allele<br />
Di qui l’ipotesi che qualcosa nel gene sia<br />
cambiato durante le generazioni.<br />
La mutazione è dinamica<br />
18
Identificazione della mutazione e<br />
clonaggio di FMR1<br />
ripetizione CGG<br />
5-50<br />
Normale<br />
polimorfismo<br />
stabile in meiosi<br />
50-200<br />
Premutazione sia nei maschi che nelle femmine<br />
instabile anche se non compromette la funzione<br />
del gene<br />
>200 Mutazione completa<br />
19
Quando avviene l’espansione<br />
•Nella meiosi, soprattutto femminile<br />
•Infatti sembra che la conversione da premutazione a<br />
mutazione avvenga nella gametogenesi femminile e non<br />
avvenga in quella maschile<br />
•(nella corea di Huntington l’espansione è nella gametogenesi maschile)<br />
•Nella mitosi, femminile e maschile (esiste un mosaicismo<br />
somatico nei tessuti e nel tempo)<br />
20
FMR1<br />
polimorfismi<br />
Il gene e’ composto di 17 esoni<br />
ed e’ lungo 38 Kb.All’interno del<br />
primo esone al 5’UTR e’ presente<br />
una serie di CGG.Nella popolazione<br />
questa regione e’ polimorfica: varia<br />
da 7 a 52 ripetizioni: l’allele piu’<br />
comune e’ quello di 30 (effetto<br />
fondatore).<br />
Gli alleli premutati sono compresi<br />
fra 60 e 200. Gli alleli<br />
completamente mutati possono<br />
raggiungere le centinaia di ripetizioni<br />
Quando il numero delle <strong>tripl</strong>ette<br />
supera 230 l’intera regione viene<br />
ipermetilata aggiungendo un gruppo<br />
metilico a C su entrambe le eliche<br />
il promotore del gene viene inattivato e FRM1 diviene silente<br />
Inoltre la cromatina non riesce a condensare correttamente<br />
21
Fenotipo di maschi e femmine con mutazione<br />
•I maschi con la mutazione manifestano il<br />
fenotipo<br />
•Le femmmine: hanno un fenotipo piu’ lieve,<br />
•1) sono 46,XX (quindi hanno un allele che produce la<br />
proteina normale)<br />
•2) Inattivazione casuale della X<br />
22
X fragile: diagnosi molecolare<br />
La PCR permette di identificare gli alleli normali e le<br />
premutazioni, ma per le espansioni maggiori, la presenza di un<br />
elevato numero di GC (la <strong>tripl</strong>etta e’ CGG) rende meno<br />
efficiente la tecnica.<br />
Si ricorre al Southern Blot dopo doppia digestione con enzimi<br />
sensibili alla metilazione: la regione espansa fino a mutazione<br />
completa e’ ipermetilata e l’enzima non taglia.<br />
23
Analisi delle mutazioni:PCR<br />
Primer<br />
Normale<br />
5-50<br />
Premutazione<br />
50-200<br />
Mutazione<br />
completa<br />
>200<br />
24
Meccanismo di formazione dell’espansione<br />
25
Distrofia miotonica<br />
26
(AD) ~ 1/20000<br />
Distrofia miotonica<br />
Fenotipo:<br />
problemi alla muscolatura scheletrica, liscia, oculare<br />
(facies caratteristica,difficolta’ ad utilizzare gli utensili,<br />
i muscoli si contraggono facilmente ma si rilassano con<br />
difficoltà)<br />
cataratta,<br />
difetti cardiaci (pace maker, terapia farmacologica)<br />
Intelletto: normale o lievemente deficitario<br />
Anomalie del sistema endocrino (spesso i pazienti sono<br />
diabetici)<br />
Aspettativa di vita ridotta<br />
27
Distrofia miotonica<br />
(AD) ~ 1/20000<br />
DMPK (mappata sul cromosoma 19) è una Ser-thr Kinasi<br />
-E’ espressa in cervello, muscolo, cuore<br />
- Il cui ruolo non è chiaro(regolazione componenti del citoscheletro,<br />
omeostasi del calcio)<br />
-Contiene (CTG)n al 3’UTR,<br />
-Nei pazienti sembra esserci un deficit di questa proteina<br />
Lieve diagnosi 20-60 anni, n=50-150, cataratta, miotonia lieve<br />
Classica diagnosi <strong>10</strong>-35 anni, n=<strong>10</strong>0-1500, muscoli, cuore, occhi,<br />
intelletto<br />
Congenita diagnosi entro i <strong>10</strong> anni, generalmente alla nascita, pochi<br />
movimenti fetali, grave ipotonia, MR nel 50% dei casi n=<strong>10</strong>00-2000, di<br />
solito ereditano l’allele espanso dalla madre,<br />
28
Il gene DMPK<br />
29
Il gene DMPK<br />
Nei pazienti (CTG)n è espanso<br />
30
Distrofia miotonica<br />
questo non e’ sufficiente per dire che quel gene e’ il<br />
responsabile della malattia: bisogna individuare anche affetti con<br />
mutazioni puntiformi.<br />
Nel caso di FMR1 ci sono affetti senza espansione con<br />
mutazioni puntiformi che impediscono l’espressione.<br />
si ritiene che il gene identificato sia il principale, ma non si<br />
e’ ancora compreso perche l’espansione provochi la patologia.<br />
Forse altera l’espressione dei geni adiacenti. Esistono modelli<br />
murini che sembrerebbero validare questa ipotesi.<br />
Ma allora bisogna ritenere DM una sindrome da geni contigui<br />
31
La diagnosi molecolare<br />
Distrofia Miotonica<br />
32
Il materiale didattico e’ presente in rete:<br />
http://www.biologia.uniba.it/DIGEMI/Didattica.html<br />
NON sono dispense, ma un ausilio allo studio sul<br />
libro<br />
33