

Annual Report 2006
Annual Report 2006
Annual Report 2006

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Analysis of Diversity in the<br />
Genus by Comparative<br />
Genomics Approach<br />
The high-quality sequence of the rice<br />
genome based on the japonica cultivar<br />
Nipponbare was revealed in 2004. Since then,<br />
the information derived from the genome<br />
sequence has been widely used in<br />
understanding the biology of rice including the<br />
identification and functional characterization of<br />
novel genes for agricultural traits. As a model<br />
cereal crop, the rice genome sequence could<br />
also be used in clarifying how the genus <br />
originated from the ancestral cereal genome, as<br />
well as how it evolved and diversified. This<br />
would not only elucidate the lineage between<br />
cultivated rice varieties from their wild<br />
relatives but would also be useful in searching<br />
for new beneficial alleles of the genes in the<br />
wild rice species that had been lost during the<br />
establishment of modern varieties. A comparative<br />
genomics approach is becoming more and more<br />
important because the wild rice has been<br />
recognized as one of the major source for novel<br />
gene alleles. Many agriculturally important<br />
genes such as disease resistance genes have<br />
been found in wild rice and introduced into the<br />
modern rice varieties.<br />
We investigated the nucleotide sequence<br />
variation among wild rice species using 900<br />
ESTs mapped at intervals of approximately 500<br />
kb throughout the genome. Primers were<br />
designed mainly from the 3-UTR region of the<br />
EST sequences and used for PCR amplification<br />
of DNA from 45 accessions belonging to 21<br />
Oryza species from the germplasm collection of<br />
NIAS and the National Institute of Genetics<br />
(NIG). The overall efficiency of amplification for<br />
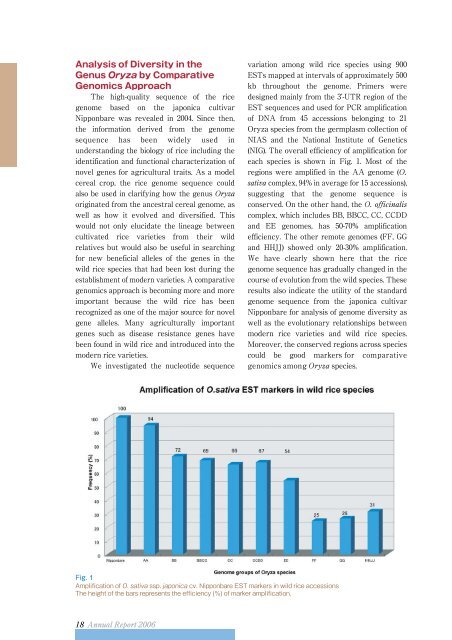
each species is shown in Fig. 1. Most of the<br />
regions were amplified in the AA genome (<br />
complex, 94% in average for 15 accessions),<br />
suggesting that the genome sequence is<br />
conserved. On the other hand, the <br />
complex, which includes BB, BBCC, CC, CCDD<br />
and EE genomes, has 50-70% amplification<br />
efficiency. The other remote genomes (FF, GG<br />
and HHJJ) showed only 20-30% amplification.<br />
We have clearly shown here that the rice<br />
genome sequence has gradually changed in the<br />
course of evolution from the wild species. These<br />
results also indicate the utility of the standard<br />
genome sequence from the japonica cultivar<br />
Nipponbare for analysis of genome diversity as<br />
well as the evolutionary relationships between<br />
modern rice varieties and wild rice species.<br />
Moreover, the conserved regions across species<br />
could be good markers for comparative<br />
genomics among species.<br />
Fig. 1<br />
Amplification of ssp. cv. Nipponbare EST markers in wild rice accessions<br />
The height of the bars represents the efficiency (%) of marker amplification.










