

Undergrad_Book_16-18_Pge_View_Print_no print marks_compressed

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
<strong>Undergrad</strong>uate Research at UMass Dartmouth<br />
depend on interactions between their side chains<br />
while the cyclic dipeptides have increased interactions<br />
between their cyclized backbones.<br />
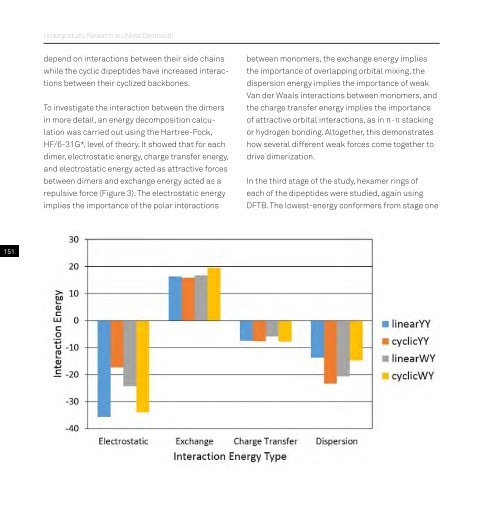
To investigate the interaction between the dimers<br />
in more detail, an energy decomposition calculation<br />
was carried out using the Hartree-Fock,<br />
HF/6-31G*, level of theory. It showed that for each<br />
dimer, electrostatic energy, charge transfer energy,<br />
and electrostatic energy acted as attractive forces<br />
between dimers and exchange energy acted as a<br />
repulsive force (Figure 3). The electrostatic energy<br />
implies the importance of the polar interactions<br />
between mo<strong>no</strong>mers, the exchange energy implies<br />
the importance of overlapping orbital mixing, the<br />
dispersion energy implies the importance of weak<br />
Van der Waals interactions between mo<strong>no</strong>mers, and<br />
the charge transfer energy implies the importance<br />
of attractive orbital interactions, as in π-π stacking<br />
or hydrogen bonding. Altogether, this demonstrates<br />
how several different weak forces come together to<br />
drive dimerization.<br />
In the third stage of the study, hexamer rings of<br />
each of the dipeptides were studied, again using<br />
DFTB. The lowest-energy conformers from stage one<br />
151
