

PDF Download - Laborwelt
PDF Download - Laborwelt
PDF Download - Laborwelt

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
B L I T Z L I C H T<br />
werden konnte, daß durch noch längere Oligonukleotide<br />
die Spezifität nicht erhöht wird.<br />
In einem theoretischen Versuch wurden für<br />
90 zufällig aus Saccharomyces cerevisiae ausgewählte<br />
Gene nicht sequenzüberlappende Oligonukleotide<br />
verschiedener Länge berechnet.<br />
Die Sequenzen der gefundenen Oligonukleotide<br />
wurden dann mittels eines Smith-<br />
Waterman-Alignments mit allen offenen Leserastern<br />
(open reading frames, ORF) der<br />
Hefe verglichen. Dabei wurde überprüft, wie<br />
oft die Sequenz eines einzelnen Oligonukleotids<br />
bestimmter Länge nochmals im Hefegenom<br />
mit einer definierten Ähnlichkeit gefunden<br />
werden kann. Die dabei ermittelte Anzahl<br />
der Treffer wurde durch die Anzahl der<br />
Oligonukleotide der gegebenen Länge dividiert,<br />
um so der Tatsache Rechnung zu tragen,<br />
daß für ein gegebenes Gen zahlenmäßig<br />
mehr kürzere als längere Oligonukleotide<br />
entworfen werden können und somit auch<br />
eine größere Zahl von Treffern gelandet werden<br />
kann. Dem Experiment wird die Beobachtung<br />
zugrundegelegt, daß zwei Transkripte<br />
mit einer Ähnlichkeit größer gleich 75%<br />
über 50 bp nicht mehr durch Hybridisierung<br />
auf einem Oligonukleotid- oder cDNA-Array<br />
voneinander unterschieden werden können 5 .<br />
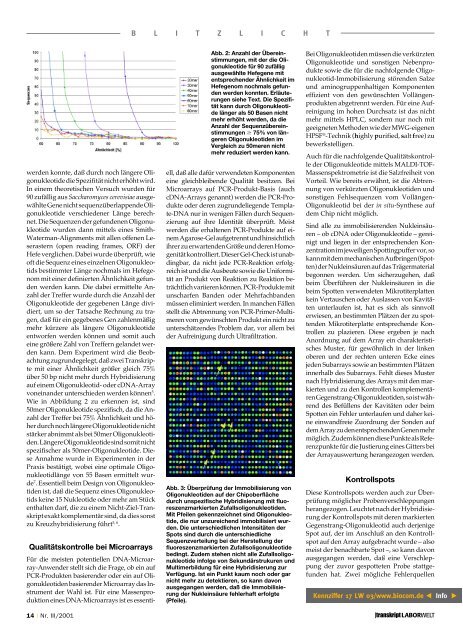
Wie in Abbildung 2 zu erkennen ist, sind<br />
50mer Oligonukleotide spezifisch, da die Anzahl<br />
der Treffer bei 75% Ähnlichkeit und höher<br />
durch noch längere Oligonukleotide nicht<br />
stärker abnimmt als bei 50mer Oligonukleotiden.<br />
Längere Oligonukleotide sind somit nicht<br />
spezifischer als 50mer-Oligonukleotide. Diese<br />
Annahme wurde in Experimenten in der<br />
Praxis bestätigt, wobei eine optimale Oligonukleotidlänge<br />
von 55 Basen ermittelt wurde<br />
7 . Essentiell beim Design von Oligonukleotiden<br />
ist, daß die Sequenz eines Oligonukleotids<br />
keine 15 Nukleotide oder mehr am Stück<br />
enthalten darf, die zu einem Nicht-Ziel-Transkript<br />
exakt komplementär sind, da dies sonst<br />
zu Kreuzhybridisierung führt 5, 6 .<br />
Qualitätskontrolle bei Microarrays<br />
Abb. 2: Anzahl der Übereinstimmungen,<br />
mit der die Oligonukleotide<br />
für 90 zufällig<br />
ausgewählte Hefegene mit<br />
entsprechender Ähnlichkeit im<br />
Hefegenom nochmals gefunden<br />
werden konnten. Erläuterungen<br />
siehe Text. Die Spezifität<br />
kann durch Oligonukleotide<br />
länger als 50 Basen nicht<br />
mehr erhöht werden, da die<br />
Anzahl der Sequenzübereinstimmungen<br />
75% von längeren<br />
Oligonukleotiden im<br />
Vergleich zu 50meren nicht<br />
mehr reduziert werden kann.<br />
Für die meisten potentiellen DNA-Microarray-Anwender<br />
stellt sich die Frage, ob ein auf<br />
PCR-Produkten basierender oder ein auf Oligonukleotiden<br />
basierender Microarray das Instrument<br />
der Wahl ist. Für eine Massenproduktion<br />
eines DNA-Microarrays ist es essentiell,<br />
daß alle dafür verwendeten Komponenten<br />
eine gleichbleibende Qualität besitzen. Bei<br />
Microarrays auf PCR-Produkt-Basis (auch<br />
cDNA-Arrays genannt) werden die PCR-Produkte<br />
oder deren zugrundeliegende Template-DNA<br />
nur in wenigen Fällen durch Sequenzierung<br />
auf ihre Identität überprüft. Meist<br />
werden die erhaltenen PCR-Produkte auf einem<br />
Agarose-Gel aufgetrennt und hinsichtlich<br />
ihrer zu erwartenden Größe und deren Homogenität<br />
kontrolliert. Dieser Gel-Check ist unabdingbar,<br />
da nicht jede PCR-Reaktion erfolgreich<br />
ist und die Ausbeute sowie die Uniformität<br />
an Produkt von Reaktion zu Reaktion beträchtlich<br />
variieren können. PCR-Produkte mit<br />
unscharfen Banden oder Mehrfachbanden<br />
müssen eliminiert werden. In manchen Fällen<br />
stellt die Abtrennung von PCR-Primer-Multimeren<br />
vom gewünschten Produkt ein nicht zu<br />
unterschätzendes Problem dar, vor allem bei<br />
der Aufreinigung durch Ultrafiltration.<br />
Abb. 3: Überprüfung der Immobilisierung von<br />
Oligonukleotiden auf der Chipoberfläche<br />
durch unspezifische Hybridisierung mit fluoreszenzmarkierten<br />
Zufallsoligonukleotiden.<br />
Mit Pfeilen gekennzeichnet sind Oligonukleotide,<br />
die nur unzureichend immobilisiert wurden.<br />
Die unterschiedlichen Intensitäten der<br />
Spots sind durch die unterschiedliche<br />
Sequenzverteilung bei der Herstellung der<br />
fluoreszenzmarkierten Zufallsoligonukleotide<br />
bedingt. Zudem stehen nicht alle Zufallsoligonukleotide<br />
infolge von Sekundärstrukuren und<br />
Multimerbildung für eine Hybridisierung zur<br />
Verfügung. Ist ein Punkt kaum noch oder gar<br />
nicht mehr zu detektieren, so kann davon<br />
ausgegangen werden, daß die Immobilisierung<br />
der Nukleinsäure fehlerhaft erfolgte<br />
(Pfeile).<br />
Bei Oligonukleotiden müssen die verkürzten<br />
Oligonukleotide und sonstigen Nebenprodukte<br />
sowie die für die nachfolgende Oligonukleotid-Immobilisierung<br />
störenden Salze<br />
und aminogruppenhaltigen Komponenten<br />
effizient von den gewünschten Vollängenprodukten<br />
abgetrennt werden. Für eine Aufreinigung<br />
im hohen Durchsatz ist das nicht<br />
mehr mittels HPLC, sondern nur noch mit<br />
geeigneten Methoden wie der MWG-eigenen<br />
HPSF ® -Technik (highly purified, salt free) zu<br />
bewerkstelligen.<br />
Auch für die nachfolgende Qualitätskontrolle<br />
der Oligonukleotide mittels MALDI-TOF-<br />
Massenspektrometrie ist die Salzfreiheit von<br />
Vorteil. Wie bereits erwähnt, ist die Abtrennung<br />
von verkürzten Oligonukleotiden und<br />
sonstigen Fehlsequenzen vom Vollängen-<br />
Oligonukleotid bei der in situ-Synthese auf<br />
dem Chip nicht möglich.<br />
Sind alle zu immobilisierenden Nukleinsäuren<br />
– ob cDNA oder Oligonukleotide – gereinigt<br />
und liegen in der entsprechenden Konzentration<br />
im jeweiligen Spottingpuffer vor, so<br />
kann mit dem mechanischen Aufbringen (Spotten)<br />
der Nukleinsäuren auf das Trägermaterial<br />
begonnen werden. Um sicherzugehen, daß<br />
beim Überführen der Nukleinsäuren in die<br />
beim Spotten verwendeten Mikrotiterplatten<br />
kein Vertauschen oder Auslassen von Kavitäten<br />
unterlaufen ist, hat es sich als sinnvoll<br />
erwiesen, an bestimmten Plätzen der zu spottenden<br />
Mikrotiterplatte entsprechende Kontrollen<br />
zu plazieren. Diese ergeben je nach<br />
Anordnung auf dem Array ein charakteristisches<br />
Muster, für gewöhnlich in der linken<br />
oberen und der rechten unteren Ecke eines<br />
jeden Subarrays sowie an bestimmten Plätzen<br />
innerhalb des Subarrays. Fehlt dieses Muster<br />
nach Hybridisierung des Arrays mit den markierten<br />
und zu den Kontrollen komplementären<br />
Gegenstrang-Oligonukleotiden, so ist während<br />
des Befüllens der Kavitäten oder beim<br />
Spotten ein Fehler unterlaufen und daher keine<br />
einwandfreie Zuordnung der Sonden auf<br />
dem Array zu den entsprechenden Genen mehr<br />
möglich. Zudem können diese Punkte als Referenzpunkte<br />
für die Justierung eines Gitters bei<br />
der Arrayauswertung herangezogen werden.<br />
Kontrollspots<br />
Diese Kontrollspots werden auch zur Überprüfung<br />
möglicher Probenverschleppungen<br />
herangezogen. Leuchtet nach der Hybridisierung<br />
der Kontrollspots mit deren markierten<br />
Gegenstrang-Oligonukleotid auch derjenige<br />
Spot auf, der im Anschluß an den Kontrollspot<br />
auf den Array aufgebracht wurde – also<br />
meist der benachbarte Spot –, so kann davon<br />
ausgegangen werden, daß eine Verschleppung<br />
der zuvor gespotteten Probe stattgefunden<br />
hat. Zwei mögliche Fehlerquellen<br />
Kennziffer 17 LW 03/www.biocom.de Info <br />
14 | Nr. III/2001 |transkript LABORWELT













