- Page 3:
Daniel John RigdenEditorFrom Protei
- Page 8 and 9:
ContentsSection I Generating and In
- Page 10 and 11:
Contentsxi4.2.1 Alpha-Helical Bundl
- Page 12 and 13:
Contentsxiii7.4.2 Predicting Bindin
- Page 14 and 15:
Contentsxv12.3 Accuracy and Added V
- Page 16 and 17:
4 J. Lee et al.about 5.3 million pr
- Page 18 and 19:
6 J. Lee et al.Table 1.1 A list of
- Page 20 and 21:
8 J. Lee et al.2000; Sorin and Pand
- Page 22 and 23:
10 J. Lee et al.Although most knowl
- Page 24 and 25:
12 J. Lee et al.Fig. 1.3 Two exampl
- Page 26 and 27:
14 J. Lee et al.rugged containing m
- Page 28 and 29:
16 J. Lee et al.1.3.4 Mathematical
- Page 30 and 31:
18 J. Lee et al.potentials, each re
- Page 32 and 33:
20 J. Lee et al.viewpoint of struct
- Page 34 and 35:
22 J. Lee et al.Hsieh MJ, Luo R (20
- Page 36 and 37:
24 J. Lee et al.Sippl MJ (1990) Cal
- Page 38 and 39:
Chapter 2Fold RecognitionLawrence A
- Page 40 and 41:
2 Fold Recognition 29It has long be
- Page 42 and 43:
2 Fold Recognition 31Fig. 2.2 Graph
- Page 44 and 45:
2 Fold Recognition 33distribute the
- Page 46 and 47:
2 Fold Recognition 35Table 2.1 (a)
- Page 48 and 49:
2 Fold Recognition 37dependent on t
- Page 50 and 51:
2 Fold Recognition 39based on the r
- Page 52 and 53:
2 Fold Recognition 41The idea of co
- Page 54 and 55:
2 Fold Recognition 43query sequence
- Page 56 and 57:
2 Fold Recognition 452.3.4 Consensu
- Page 58 and 59:
2 Fold Recognition 472.4 Alignment
- Page 60 and 61:
2 Fold Recognition 49statistical me
- Page 62 and 63:
2 Fold Recognition 51methods (e.g.
- Page 64 and 65:
2 Fold Recognition 53Berman HM, Wes
- Page 66 and 67:
2 Fold Recognition 55Tress ML, Jone
- Page 68 and 69:
58 A. Fiserwith more than 50% seque
- Page 70 and 71:
60 A. FiserIn contrast to ab initio
- Page 72 and 73:
62 A. FiserTable 3.1 (continued)SWI
- Page 74 and 75:
64 A. Fiserbetween fold recognition
- Page 76 and 77: 66 A. FiserFig. 3.1 Comparing accur
- Page 78 and 79: 68 A. Fiserinto conserved core regi
- Page 80 and 81: 70 A. Fiseralignments are built and
- Page 82 and 83: 72 A. Fiserfold, such as the hyperv
- Page 84 and 85: 74 A. Fiserfunctions delivers impro
- Page 86 and 87: 76 A. Fiserfrom efforts of genome s
- Page 88 and 89: 78 A. FiserA rigorous statistical e
- Page 90 and 91: 80 A. Fiser(Clore et al. 1993). Cha
- Page 92 and 93: 82 A. FiserImproved and new methods
- Page 94 and 95: 84 A. FiserClaessens M, Van Cutsem
- Page 96 and 97: 86 A. FiserHavel TF, Snow ME (1991)
- Page 98 and 99: 88 A. FiserPetrey D, Xiang Z, Tang
- Page 100 and 101: 90 A. Fiservan Vlijmen HW, Karplus
- Page 102 and 103: 92 T. Nugent and D.T. Jones4.2 Stru
- Page 104 and 105: 94 T. Nugent and D.T. JonesFig. 4.2
- Page 106 and 107: 96 T. Nugent and D.T. JonesTable 4.
- Page 108 and 109: 98 T. Nugent and D.T. Jones2000), d
- Page 110 and 111: 100 T. Nugent and D.T. JonesTable 4
- Page 112 and 113: 102 T. Nugent and D.T. JonesFig. 4.
- Page 114 and 115: 104 T. Nugent and D.T. JonesFig. 4.
- Page 116 and 117: 106 T. Nugent and D.T. Jonessubdivi
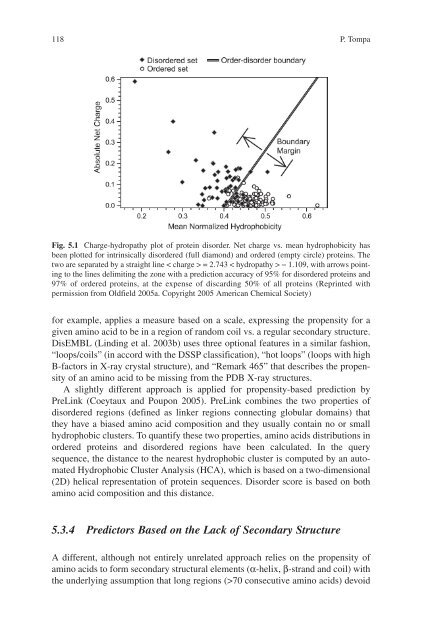
- Page 118 and 119: 108 T. Nugent and D.T. Jonescomplex
- Page 120 and 121: 110 T. Nugent and D.T. JonesMartell
- Page 122 and 123: Chapter 5Bioinformatics Approaches
- Page 124 and 125: 5 Structure and Function of Intrins
- Page 128 and 129: 5 Structure and Function of Intrins
- Page 130 and 131: 5 Structure and Function of Intrins
- Page 132 and 133: 5 Structure and Function of Intrins
- Page 134 and 135: 5 Structure and Function of Intrins
- Page 136 and 137: 5 Structure and Function of Intrins
- Page 138 and 139: 5 Structure and Function of Intrins
- Page 140 and 141: 5 Structure and Function of Intrins
- Page 142 and 143: 5 Structure and Function of Intrins
- Page 144 and 145: 5 Structure and Function of Intrins
- Page 146 and 147: 5 Structure and Function of Intrins
- Page 148 and 149: 5 Structure and Function of Intrins
- Page 150 and 151: Chapter 6Function Diversity Within
- Page 152 and 153: 6 Function Diversity Within Folds a
- Page 154 and 155: 6 Function Diversity Within Folds a
- Page 156 and 157: 6 Function Diversity Within Folds a
- Page 158 and 159: 6 Function Diversity Within Folds a
- Page 160 and 161: 6 Function Diversity Within Folds a
- Page 162 and 163: 6 Function Diversity Within Folds a
- Page 164 and 165: 6 Function Diversity Within Folds a
- Page 166 and 167: 6 Function Diversity Within Folds a
- Page 168 and 169: 6 Function Diversity Within Folds a
- Page 170 and 171: 6 Function Diversity Within Folds a
- Page 172 and 173: 6 Function Diversity Within Folds a
- Page 174 and 175: Chapter 7Predicting Protein Functio
- Page 176 and 177:
7 Predicting Protein Function from
- Page 178 and 179:
7 Predicting Protein Function from
- Page 180 and 181:
7 Predicting Protein Function from
- Page 182 and 183:
7 Predicting Protein Function from
- Page 184 and 185:
7 Predicting Protein Function from
- Page 186 and 187:
7 Predicting Protein Function from
- Page 188 and 189:
7 Predicting Protein Function from
- Page 190 and 191:
Table 7.1 Online resources and tool
- Page 192 and 193:
7 Predicting Protein Function from
- Page 194 and 195:
Chapter 83D MotifsElaine C. Meng, B
- Page 196 and 197:
8 3D Motifs 189clustering similar s
- Page 198 and 199:
8 3D Motifs 191To improve the signa
- Page 200 and 201:
8 3D Motifs 193structures together.
- Page 202 and 203:
8 3D Motifs 195Table 8.1 (continued
- Page 204 and 205:
8 3D Motifs 1978.3.1 User-Defined M
- Page 206 and 207:
8 3D Motifs 199Fig. 8.3 The FFF mot
- Page 208 and 209:
8 3D Motifs 2018.3.2 Motif Discover
- Page 210 and 211:
8 3D Motifs 203In addition to SITE
- Page 212 and 213:
8 3D Motifs 205folds; they shared a
- Page 214 and 215:
8 3D Motifs 207from a training set
- Page 216 and 217:
8 3D Motifs 209Table 8.3 Web server
- Page 218 and 219:
8 3D Motifs 211its greater overall
- Page 220 and 221:
8 3D Motifs 213Ideally, a 3D motif
- Page 222 and 223:
8 3D Motifs 215Ivanisenko VA, Pintu
- Page 224 and 225:
Chapter 9Protein Dynamics: From Str
- Page 226 and 227:
9 Protein Dynamics: From Structure
- Page 228 and 229:
9 Protein Dynamics: From Structure
- Page 230 and 231:
9 Protein Dynamics: From Structure
- Page 232 and 233:
9 Protein Dynamics: From Structure
- Page 234 and 235:
9 Protein Dynamics: From Structure
- Page 236 and 237:
9 Protein Dynamics: From Structure
- Page 238 and 239:
9 Protein Dynamics: From Structure
- Page 240 and 241:
9 Protein Dynamics: From Structure
- Page 242 and 243:
9 Protein Dynamics: From Structure
- Page 244 and 245:
9 Protein Dynamics: From Structure
- Page 246 and 247:
9 Protein Dynamics: From Structure
- Page 248 and 249:
9 Protein Dynamics: From Structure
- Page 250 and 251:
9 Protein Dynamics: From Structure
- Page 252 and 253:
9 Protein Dynamics: From Structure
- Page 254 and 255:
9 Protein Dynamics: From Structure
- Page 256 and 257:
9 Protein Dynamics: From Structure
- Page 258 and 259:
252 R.A. LaskowskiConsequently, man
- Page 260 and 261:
254 R.A. Laskowskiucla.edu, and Pro
- Page 262 and 263:
256 R.A. LaskowskiFig. 10.2 Gene On
- Page 264 and 265:
258 R.A. Laskowski10.2.5 Protein In
- Page 266 and 267:
260 R.A. LaskowskiFig. 10.4 Schemat
- Page 268 and 269:
262 R.A. Laskowskiaccessibility and
- Page 270 and 271:
264 R.A. LaskowskiFig. 10.6 A ligan
- Page 272 and 273:
266 R.A. LaskowskiGly97Gly99Tyr98Ty
- Page 274 and 275:
268 R.A. LaskowskiFig. 10.8 Example
- Page 276 and 277:
270 R.A. LaskowskiReferencesAltschu
- Page 278 and 279:
272 R.A. LaskowskiXenarios I, Salwi
- Page 280 and 281:
274 J.D. Watson and J.M. ThorntonTh
- Page 282 and 283:
276 J.D. Watson and J.M. ThorntonTa
- Page 284 and 285:
278 J.D. Watson and J.M. Thorntonva
- Page 286 and 287:
280 J.D. Watson and J.M. Thorntonfo
- Page 288 and 289:
282 J.D. Watson and J.M. Thorntonin
- Page 290 and 291:
284 J.D. Watson and J.M. Thorntonan
- Page 292 and 293:
286 J.D. Watson and J.M. ThorntonFi
- Page 294 and 295:
288 J.D. Watson and J.M. ThorntonPD
- Page 296 and 297:
290 J.D. Watson and J.M. ThorntonKi
- Page 298 and 299:
Chapter 12Prediction of Protein Fun
- Page 300 and 301:
12 Prediction of Protein Function f
- Page 302 and 303:
12 Prediction of Protein Function f
- Page 304 and 305:
12 Prediction of Protein Function f
- Page 306 and 307:
12 Prediction of Protein Function f
- Page 308 and 309:
12 Prediction of Protein Function f
- Page 310 and 311:
12 Prediction of Protein Function f
- Page 312 and 313:
12 Prediction of Protein Function f
- Page 314 and 315:
12 Prediction of Protein Function f
- Page 316 and 317:
12 Prediction of Protein Function f
- Page 318 and 319:
12 Prediction of Protein Function f
- Page 320 and 321:
12 Prediction of Protein Function f
- Page 322 and 323:
12 Prediction of Protein Function f
- Page 324 and 325:
320 IndexConsensus templates, 205Co
- Page 326 and 327:
322 IndexLigand-binding templates,
- Page 328:
324 IndexStructure-function linkage