

Querschnitt 21 / Februar 2007 - h_da: Hochschule Darmstadt
Querschnitt 21 / Februar 2007 - h_da: Hochschule Darmstadt
Querschnitt 21 / Februar 2007 - h_da: Hochschule Darmstadt

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
<strong>Querschnitt</strong> <strong>21</strong><br />
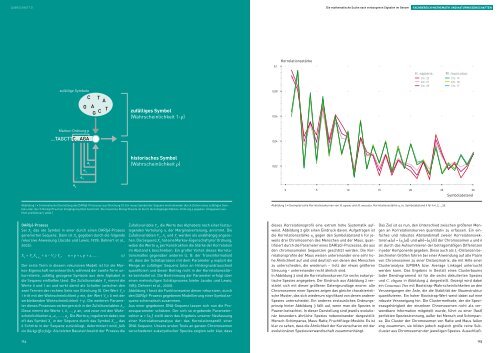
Dar(p)-prozess<br />
Sei X n <strong>da</strong>s nte Symbol in einer durch einen DAr(p)-Prozess<br />
generierten Sequenz. Dann ist X n gegeben durch die folgende<br />
rekursive Anweisung (Jacobs und Lewis, 1978; Dehnert et al.,<br />
2003):<br />
X n = V n X n-An + (1 – V n ) Y n , n = p + 1, p + 2, … . (1)<br />
Der erste Term in diesem rekursiven Modell ist für die Markov-eigenschaft<br />
verantwortlich, während der zweite Term unkorrelierte,<br />
zufällig gezogene Symbole aus dem Alphabet in<br />
die Sequenz einfließen lässt. Die Zufallsvariable V n nimmt die<br />
Werte 0 und 1 an und wirkt <strong>da</strong>mit als Schalter zwischen den<br />
zwei Termen der rechten Seite von Gleichung (1). Der Wert V n =<br />
1 tritt mit der Wahrscheinlichkeit ρ ein, der Wert V n = 0 mit der<br />
verbleibenden Wahrscheinlichkeit 1–ρ. Die weiteren Parameter<br />
dieses Prozesses verbergen sich in der Zufallsvariablen A n .<br />
Diese nimmt die Werte 1, 2, …, p an, und zwar mit den Wahrscheinlichkeiten<br />
α 1 , α 2 , …. , α p . Die Werte α k regulieren <strong>da</strong>bei, wie<br />
oft <strong>da</strong>s Symbol X n in der Sequenz durch <strong>da</strong>s Symbol X n-k , <strong>da</strong>s<br />
k Schritte in der Sequenz zurückliegt, determiniert wird, falls<br />
ein Rückgriff erfolgt. Als letzten Baustein besitzt der Prozess die<br />
11<br />
zufällige Symbole<br />
Markov-Ordnung p<br />
…TAGCTTC…AGA<br />
. . .<br />
α p<br />
α 3<br />
C T<br />
A<br />
G A<br />
G C<br />
T<br />
α 2<br />
α 1<br />
zufälliges Symbol<br />
(Wahrscheinlichkeit 1-ρ)<br />
historisches Symbol<br />
(Wahrscheinlichkeit ρ)<br />
Abbildung 1 • Schematische Darstellung des DAr(p)-Prozesses aus Gleichung (1). ein neues Symbol der Sequenz wird entweder durch Ziehen eines zufälligen Symbols<br />
oder durch rückgriff auf ein Vorgängersymbol bestimmt. Die maximale rückgriffweite ist durch die festgelegte Markov-Ordnung p gegeben. (Angepasst aus:<br />
Hütt und Dehnert, 2006.)<br />
Zufallsvariable Y n , die Werte des Alphabets nach einer festzulegenden<br />
Verteilung π, der Marginalverteilung, annimmt. Die<br />
Zufallsvariablen V n , A n und Y n werden als unabhängig angesehen.<br />
Die Sequenz X n hat eine Markov-eigenschaft pter Ordnung,<br />
wobei die Werte α k per Konstruktion die Stärke der Korrelation<br />
im Abstand k beschreiben. ein großer Vorteil dieses Korrelationsmaßes<br />
gegenüber anderen (z. B. der Transinformation)<br />
ist, <strong>da</strong>ss der Schätzprozess mit dem Parameter ρ explizit die<br />
Menge an zufälliger Sequenz (also an Hintergrundrauschen)<br />
quantifiziert und dieser Beitrag nicht in der Korrelationsstärke<br />
beinhaltet ist. Die Bestimmung der Parameter erfolgt über<br />
einen mehrstufigen Schätzprozess (siehe Jacobs und Lewis,<br />
1983; Dehnert et al., 2006).<br />
Abbildung 1 fasst die Funktionsweise dieser rekursiven, durch<br />
den DAr(p)-Prozess gegebenen Modellierung einer Symbolsequenz<br />
schematisch zusammen.<br />
Aus einer gegebenen DNA-Sequenz lassen sich nun die Prozessparameter<br />
schätzen. Der sich so ergebende Parametervektor<br />
α = {α k } stellt <strong>da</strong>nn <strong>da</strong>s ergebnis unserer Neufassung<br />
einer Korrelationsanalyse <strong>da</strong>r: <strong>da</strong>s Korrelationsprofil einer<br />
DNA-Sequenz. unsere ersten Tests an ganzen Chromosomen<br />
verschiedener eukaryotischer Spezies zeigten sehr klar, <strong>da</strong>ss<br />
0,1<br />
0,08<br />
0,06<br />
0,04<br />
0,02<br />
Korrelationsstärke<br />
Die mathematische suche nach verborgenen signalen im genom<br />
H. sapiens:<br />
Chr. 22<br />
Chr. <strong>21</strong><br />
Chr. 20<br />
M. musculus:<br />
Chr. 19<br />
Chr. 18<br />
Chr. 17<br />
0 5<br />
10 15 20<br />
25 30<br />
Abbildung 2 • exemplarische Korrelationskurven von H. sapiens und M. musculus. Korrelationsstärke α k vs. Symbolabstand k für k=1,2,…,30.<br />
dieses Korrelationsprofil eine extrem hohe Systematik aufweist.<br />
Abbildung 2 gibt einen eindruck <strong>da</strong>von. Aufgetragen ist<br />
die Korrelationsstärke α k gegen den Symbolabstand k für jeweils<br />
drei Chromosomen des Menschen und der Maus, quantifiziert<br />
durch die Parameter eines DAr(30)-Prozesses, die aus<br />
den chromosomalen Sequenzen geschätzt werden. Die Korrelationsprofile<br />
der Maus weisen untereinander eine sehr hohe<br />
Ähnlichkeit auf und sind deutlich von denen des Menschen<br />
zu unterscheiden, die wiederum – trotz der etwas größeren<br />
Streuung – untereinander recht ähnlich sind.<br />
In Abbildung 3 sind die Korrelationskurven für sechs eukaryotische<br />
Spezies angegeben. Der eindruck aus Abbildung 2 verstärkt<br />
sich mit dieser größeren Datengrundlage enorm: alle<br />
Chromosomen einer Spezies zeigen <strong>da</strong>s gleiche charakteristische<br />
Muster, <strong>da</strong>s sich wiederum signifikant von denen anderer<br />
Spezies unterscheidet. ein anderes erstaunliches Ordnungsprinzip<br />
hinter Abbildung 3 fällt auf, wenn man die Spezies in<br />
Paaren betrachtet. In dieser Darstellung sind jeweils evolutionär<br />
besonders ähnliche Spezies nebeneinander <strong>da</strong>rgestellt:<br />
Mensch-Schimpanse, Maus-ratte, Fruchtfliege-Moskito. es ist<br />
klar zu sehen, <strong>da</strong>ss die Ähnlichkeit der Kurvenscharen mit der<br />
evolutionären Speziesverwandtschaft zusammenhängt.<br />
FAchbereich mAthemAtik und nAturwissenschAFten<br />
Symbolabstand<br />
Das Ziel ist es nun, den unterschied zwischen größeren Mengen<br />
an Korrelationskurven quantitativ zu erfassen. ein einfaches<br />
und robustes Abstandsmaß zweier Korrelationsvektoren<br />
α(a) = {α k (a)} und α(b)={α k (b)} der Chromosomen a und b<br />
ist durch <strong>da</strong>s Aufsummieren der betragsmäßigen Differenzen<br />
in jeder Komponente gegeben. Diese auch als L 1 -Distanzen bezeichneten<br />
Größen führen bei einer Anwendung auf alle Paare<br />
von Chromosomen zu einer Distanzmatrix, die mit Hilfe einer<br />
Clusteranalyse (uPGMA bzw. Average Linkage) untersucht<br />
werden kann. Das ergebnis in Gestalt eines Clusterbaums<br />
(oder Dendrogramms) ist für die sechs diskutierten Spezies<br />
und C. elegans in Abbildung 4 <strong>da</strong>rgestellt. Gezeigt wird <strong>da</strong>bei<br />
ein Consensus Tree mit Bootstrap-Wahrscheinlichkeiten an den<br />
Verzweigungen der Äste, die die Stabilität der Baumstruktur<br />
quantifizieren. ein hoher Bootstrap-Wert weist <strong>da</strong>bei auf eine<br />
robuste Verzweigung hin. Die Clustermethode, der die Spezieszugehörigkeit<br />
der einzelnen Chromosomen nicht als verwendbare<br />
Information mitgeteilt wurde, führt zu einer (fast)<br />
perfekten Speziestrennung, außer bei Mensch und Schimpanse.<br />
Die Cluster der Chromosomen von ratte und Maus fallen<br />
eng zusammen, sie bilden jedoch zugleich große reine Subcluster<br />
aus Chromosomen der jeweiligen Spezies. Ausschließ-<br />
115


