

PDF Download - Laborwelt
PDF Download - Laborwelt
PDF Download - Laborwelt

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
Blitzlicht Genomforschung<br />
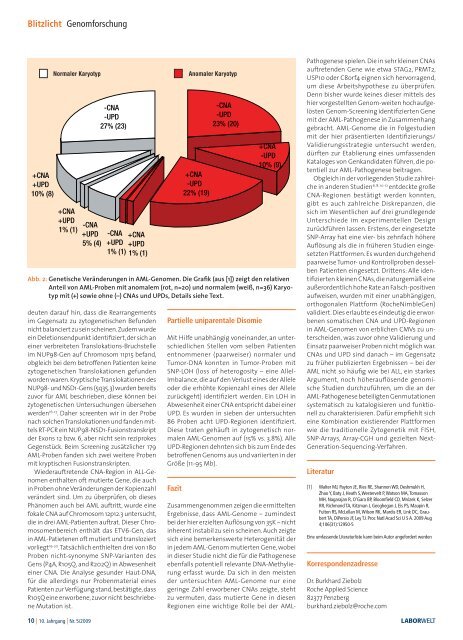
+CNA<br />
+UPD<br />
10% (8)<br />
Normaler Karyotyp Anomaler Karyotyp<br />
+CNA<br />
+UPD<br />
1% (1) -CNA<br />
+UPD<br />
5% (4)<br />
-CNA<br />
-UPD<br />
27% (23)<br />
-CNA +CNA<br />
+UPD +UPD<br />
1% (1) 1% (1)<br />
deuten darauf hin, dass die Rearrangements<br />
im Gegensatz zu zytogenetischen Befunden<br />
nicht balanciert zu sein scheinen. Zudem wurde<br />
ein Deletionsendpunkt identifiziert, der sich an<br />
einer verbreiteten Translokations-Bruchstelle<br />
im NUP98-Gen auf Chromosom 11p15 befand,<br />
obgleich bei dem betroffenen Patienten keine<br />
zytogenetischen Translokationen gefunden<br />
worden waren. Kryptische Translokationen des<br />
NUP98- und NSD1-Gens (5q35.3) wurden bereits<br />
zuvor für AML beschrieben, diese können bei<br />
zytogenetischen Untersuchungen übersehen<br />
werden 16-17 . Daher screenten wir in der Probe<br />
nach solchen Translokationen und fanden mittels<br />
RT-PCR ein NUP98-NSD1-Fusionstranskript<br />
der Exons 12 bzw. 6, aber nicht sein reziprokes<br />
Gegenstück. Beim Screening zusätzlicher 179<br />
AML-Proben fanden sich zwei weitere Proben<br />
mit kryptischen Fusionstrans kripten.<br />
Wiederauftretende CNA-Region in ALL-Genomen<br />
enthalten oft mutierte Gene, die auch<br />
in Proben ohne Veränderungen der Kopienzahl<br />
verändert sind. Um zu überprüfen, ob dieses<br />
Phänomen auch bei AML auftritt, wurde eine<br />
fokale CNA auf Chromosom 12p12.3 untersucht,<br />
die in drei AML-Patienten auftrat. Dieser Chromosomenbereich<br />
enthält das ETV6-Gen, das<br />
in AML-Patietenen oft mutiert und transloziert<br />
vorliegt 19-20 . Tatsächlich enthielten drei von 180<br />
Proben nicht-synonyme SNP-Varianten des<br />
Gens (P4A, R105Q, and R202Q) in Abwesenheit<br />
einer CNA. Die Analyse gesunder Haut-DNA,<br />
für die allerdings nur Probenmaterial eines<br />
Patienten zur Verfügung stand, bestätigte, dass<br />
R105Q eine erworbene, zuvor nicht beschriebene<br />
Mutation ist.<br />
+CNA<br />
-UPD<br />
22% (19)<br />
-CNA<br />
-UPD<br />
23% (20)<br />
Partielle uniparentale Disomie<br />
+CNA<br />
-UPD<br />
10% (9)<br />
Abb. 2: Genetische Veränderungen in AML-Genomen. Die Grafik (aus [1]) zeigt den relativen<br />
Anteil von AML-Proben mit anomalem (rot, n=20) und normalem (weiß, n=36) Karyotyp<br />
mit (+) sowie ohne (–) CNAs und UPDs, Details siehe Text.<br />
Mit Hilfe unabhängig voneinander, an unterschiedlichen<br />
Stellen vom selben Patienten<br />
entnommener (paarweiser) normaler und<br />
Tumor-DNA konnten in Tumor-Proben mit<br />
SNP-LOH (loss of heterogosity – eine Allel-<br />
Imbalance, die auf den Verlust eines der Allele<br />
oder die erhöhte Kopienzahl eines der Allele<br />
zurückgeht) identifiziert werden. Ein LOH in<br />
Abwesenheit einer CNA entspricht dabei einer<br />
UPD. Es wurden in sieben der untersuchten<br />
86 Proben acht UPD-Regionen identifiziert.<br />
Diese traten gehäuft in zytogenetisch normalen<br />
AML-Genomen auf (15% vs. 3.8%). Alle<br />
UPD-Regionen dehnten sich bis zum Ende des<br />
betroffenen Genoms aus und variierten in der<br />
Größe (11-95 Mb).<br />
Fazit<br />
Zusammengenommen zeigen die ermittelten<br />
Ergebnisse, dass AML-Genome – zumindest<br />
bei der hier erzielten Auflösung von 35K – nicht<br />
inherent instabil zu sein scheinen. Auch zeigte<br />
sich eine bemerkenswerte Heterogenität der<br />
in jedem AML-Genom mutierten Gene, wobei<br />
in dieser Studie nicht die für die Pathogenese<br />
ebenfalls potentiell relevante DNA-Methylierung<br />
erfasst wurde. Da sich in den meisten<br />
der untersuchten AML-Genome nur eine<br />
geringe Zahl erworbener CNAs zeigte, steht<br />
zu vermuten, dass mutierte Gene in diesen<br />
Regionen eine wichtige Rolle bei der AML-<br />
Pathogenese spielen. Die in sehr kleinen CNAs<br />
auftretenden Gene wie etwa STAG2, PRMT2,<br />
USP10 oder C8orf4 eignen sich hervorragend,<br />
um diese Arbeitshypothese zu überprüfen.<br />
Denn bisher wurde keines dieser mittels des<br />
hier vorgestellten Genom-weiten hochaufgelösten<br />
Genom-Screening identifizierten Gene<br />
mit der AML-Pathogenese in Zusammenhang<br />
gebracht. AML-Genome die in Folgestudien<br />
mit der hier präsentierten Identifizierungs/<br />
Validierungsstrategie untersucht werden,<br />
dürften zur Etablierung eines umfassenden<br />
Kataloges von Genkandidaten führen, die potentiell<br />
zur AML-Pathogenese beitragen.<br />
Obgleich in der vorliegenden Studie zahlreiche<br />
in anderen Studien 6, 8, 10-12 entdeckte große<br />
CNA-Regionen bestätigt werden konnten,<br />
gibt es auch zahlreiche Diskrepanzen, die<br />
sich im Wesentlichen auf drei grundlegende<br />
Unterschiede im experimentellen Design<br />
zurückführen lassen. Erstens, der eingesetzte<br />
SNP-Array hat eine vier- bis zehnfach höhere<br />
Auflösung als die in früheren Studien eingesetzten<br />
Plattformen. Es wurden durchgehend<br />
paarweise Tumor- und Kontrollproben desselben<br />
Patienten eingesetzt. Drittens: Alle identifizierten<br />
kleinen CNAs, die naturgemäß eine<br />
außerordentlich hohe Rate an Falsch-positiven<br />
aufweisen, wurden mit einer unabhängigen,<br />
orthogonalen Plattform (RocheNimbleGen)<br />
validiert. Dies erlaubte es eindeutig die erworbenen<br />
somatischen CNA und UPD-Regionen<br />
in AML-Genomen von erblichen CMVs zu unterscheiden,<br />
was zuvor ohne Validierung und<br />
Einsatz paarweiser Proben nicht möglich war.<br />
CNAs und UPD sind danach – im Gegensatz<br />
zu früher publizierten Ergebnissen – bei der<br />
AML nicht so häufig wie bei ALL, ein starkes<br />
Argument, noch höherauflösende genomische<br />
Studien durchzuführen, um die an der<br />
AML-Pathogenese beteiligten Genmutationen<br />
systematisch zu katalogisieren und funktionell<br />
zu charakterisieren. Dafür empfiehlt sich<br />
eine Kombination existierender Plattformen<br />
wie die traditionelle Zytogenetik mit FISH,<br />
SNP-Arrays, Array-CGH und gezielten Next-<br />
Generation-Sequencing-Verfahren.<br />
Literatur<br />
[1] Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H,<br />
Zhao Y, Baty J, Heath S, Westervelt P, Watson MA, Tomasson<br />
MH, Nagarajan R, O‘Gara BP, Bloomfield CD, Mrózek K, Selzer<br />
RR, Richmond TA, Kitzman J, Geoghegan J, Eis PS, Maupin R,<br />
Fulton RS, McLellan M, Wilson RK, Mardis ER, Link DC, Graubert<br />
TA, DiPersio JF, Ley TJ. Proc Natl Acad Sci U S A. 2009 Aug<br />
4;106(31):12950-5<br />
Eine umfassende Literaturliste kann beim Autor angefordert werden<br />
Korrespondenzadresse<br />
Dr. Burkhard Ziebolz<br />
Roche Applied Science<br />
82377 Penzberg<br />
burkhard.ziebolz@roche.com<br />
10 | 10. Jahrgang | Nr. 5/2009 LABORWElT














