

PDF Download - Laborwelt
PDF Download - Laborwelt
PDF Download - Laborwelt

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
R E P O R T<br />
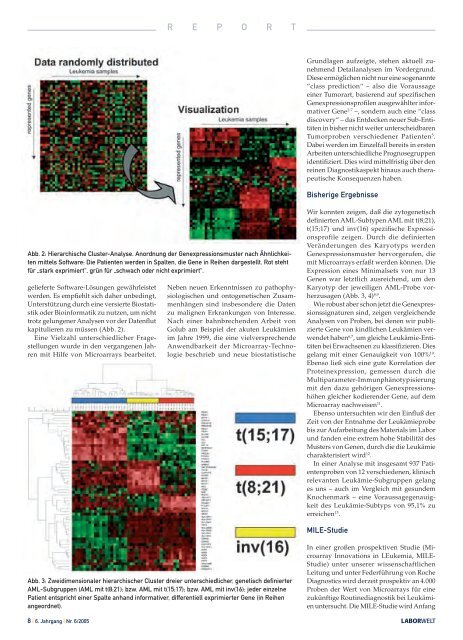
Abb. 2: Hierarchische Cluster-Analyse. Anordnung der Genexpressionsmuster nach Ähnlichkeiten<br />
mittels Software: Die Patienten werden in Spalten, die Gene in Reihen dargestellt. Rot steht<br />
für „stark exprimiert“, grün für „schwach oder nicht exprimiert“.<br />
gelieferte Software-Lösungen gewährleistet<br />
werden. Es empfiehlt sich daher unbedingt,<br />
Unterstützung durch eine versierte Biostatistik<br />
oder Bioinformatik zu nutzen, um nicht<br />
trotz gelungener Analysen vor der Datenflut<br />
kapitulieren zu müssen (Abb. 2).<br />
Eine Vielzahl unterschiedlicher Fragestellungen<br />
wurde in den vergangenen Jahren<br />
mit Hilfe von Microarrays bearbeitet.<br />
Neben neuen Erkenntnissen zu pathophysiologischen<br />
und ontogenetischen Zusammenhängen<br />
sind insbesondere die Daten<br />
zu malignen Erkrankungen von Interesse.<br />
Nach einer bahnbrechenden Arbeit von<br />
Golub am Beispiel der akuten Leukämien<br />
im Jahre 1999, die eine vielversprechende<br />
Anwendbarkeit der Microarray-Technologie<br />
beschrieb und neue biostatistische<br />
Abb. 3: Zweidimensionaler hierarchischer Cluster dreier unterschiedlicher, genetisch definierter<br />
AML-Subgruppen (AML mit t(8;21); bzw. AML mit t(15;17); bzw. AML mit inv(16); jeder einzelne<br />
Patient entspricht einer Spalte anhand informativer, differentiell exprimierter Gene (in Reihen<br />
angeordnet).<br />
Grundlagen aufzeigte, stehen aktuell zunehmend<br />
Detailanalysen im Vordergrund.<br />
Diese ermöglichen nicht nur eine sogenannte<br />
“class prediction“ – also die Voraussage<br />
einer Tumorart, basierend auf spezifischen<br />
Genexpressionsprofilen ausgewählter informativer<br />
Gene 2-7 –, sondern auch eine “class<br />
discovery“ – das Entdecken neuer Sub-Entitäten<br />
in bisher nicht weiter unterscheidbaren<br />
Tumorproben verschiedener Patienten 5 .<br />
Dabei werden im Einzelfall bereits in ersten<br />
Arbeiten unterschiedliche Prognosegruppen<br />
identifiziert. Dies wird mittelfristig über den<br />
reinen Diagnostikaspekt hinaus auch therapeutische<br />
Konsequenzen haben.<br />
Bisherige Ergebnisse<br />
Wir konnten zeigen, daß die zytogenetisch<br />
definierten AML-Subtypen AML mit t(8;21),<br />
t(15;17) und inv(16) spezifische Expressionsprofile<br />
zeigen. Durch die definierten<br />
Veränderungen des Karyotyps werden<br />
Genexpressionsmuster hervorgerufen, die<br />
mit Microarrays erfaßt werden können. Die<br />
Expression eines Minimalsets von nur 13<br />
Genen war letztlich ausreichend, um den<br />
Karyotyp der jeweiligen AML-Probe vorherzusagen<br />
(Abb. 3, 4) 8,9 .<br />
Wie robust aber schon jetzt die Genexpressionssignaturen<br />
sind, zeigen vergleichende<br />
Analysen von Proben, bei denen wir publizierte<br />
Gene von kindlichen Leukämien verwendet<br />
haben 6,7 , um gleiche Leukämie-Entitäten<br />
bei Erwachsenen zu klassifizieren. Dies<br />
gelang mit einer Genauigkeit von 100% 10 .<br />
Ebenso ließ sich eine gute Korrelation der<br />
Proteinexpression, gemessen durch die<br />
Multiparameter-Immunphänotypisierung<br />
mit den dazu gehörigen Genexpressionshöhen<br />
gleicher kodierender Gene, auf dem<br />
Microarray nachweisen 11 .<br />
Ebenso untersuchten wir den Einfluß der<br />
Zeit von der Entnahme der Leukämieprobe<br />
bis zur Aufarbeitung des Materials im Labor<br />
und fanden eine extrem hohe Stabilität des<br />
Musters von Genen, durch die die Leukämie<br />
charakterisiert wird 12 .<br />
In einer Analyse mit insgesamt 937 Patientenproben<br />
von 12 verschiedenen, klinisch<br />
relevanten Leukämie-Subgruppen gelang<br />
es uns – auch im Vergleich mit gesundem<br />
Knochenmark – eine Voraussagegenauigkeit<br />
des Leukämie-Subtyps von 95,1% zu<br />
erreichen 13 .<br />
MILE-Studie<br />
In einer großen prospektiven Studie (Microarray<br />
Innovations in LEukemia, MILE-<br />
Studie) unter unserer wissenschaftlichen<br />
Leitung und unter Federführung von Roche<br />
Diagnostics wird derzeit prospektiv an 4.000<br />
Proben der Wert von Microarrays für eine<br />
zukünftige Routinediagnostik bei Leukämien<br />
untersucht. Die MILE-Studie wird Anfang<br />
8 | 6. Jahrgang | Nr. 6/2005 LABORWELT













