

bbc 2015
BBC2015_booklet
BBC2015_booklet

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
BeNeLux Bioinformatics Conference – Antwerp, December 7-8 <strong>2015</strong><br />
Abstract ID: P<br />
Poster<br />
10th Benelux Bioinformatics Conference <strong>bbc</strong> <strong>2015</strong><br />
P42. EARLY FOLDING AND LOCAL INTERACTIONS<br />
R. Pancsa 1 , M. Varadi 1 , E. Cilia 2,3 , D. Raimondi 1,2,3 & W. F. Vranken 1,3,* .<br />
Structural Biology Research Centre, VIB and Structural Biology Brussels, Vrije Universiteit Brussel, Brussels, Belgium 1 ;<br />
Machine Learning Group, Université Libre de Bruxelles, Brussels, Belgium 2 ; Interuniversity Institute of Bioinformatics<br />
in Brussels (IB) 2 , Brussels, Belgium 3 . * wvranken@vub.ac.be<br />
INTRODUCTION<br />
Protein folding is in its early stages largely determined by<br />
the protein sequence and complex local interactions<br />
between amino acids, resulting in the formation of foldons<br />
that provide the context for further folding into the native<br />
state. These early folding processes are therefore<br />
important to understand subsequent folding steps and their<br />
influence on, for example, aggregation, but they are<br />
difficult to study experimentally. We here address this<br />
issue computationally by assembling and analysing a<br />
dataset on early folding residues from hydrogen deuterium<br />
exchange (HDX) data from NMR and MS, and analyse<br />
how they relate to the sequence-based backbone dynamics<br />
predictions from DynaMine (Cilia et al. 2013, 2014) and<br />
evolutionary information from multiple sequence<br />
alignments.<br />
METHODS<br />
We assembled a dataset of HDX experimental data from<br />
NMR and MS from literature for 57 proteins totalling<br />
4172 residues. The data was classified by the into early,<br />
intermediate and late classes depending on the folding<br />
time where protection of the backbone NH was observed,<br />
and into strong, medium and weak classes depending on<br />
how long the amides remain protected upon unfolding the<br />
native state. This resulted in 219 residue sets that are<br />
organised in XML files and loaded into a database that is<br />
made available online via http://start2fold.eu.<br />
The DynaMine predictions were run locally with a new<br />
version of the software that handles C- and N-terminal<br />
effects. These original predictions were then normalised<br />
by shifting them so that the maximum prediction value for<br />
each protein is always 1.0, so not affecting the relative<br />
differences between the prediction values within each<br />
protein, but effectively normalising the values between<br />
different proteins. MSAs were generated for each<br />
sequence in the dataset using HHblits and Jackhmmer with<br />
3 iterations and E value threshold of 10 -4 . All the retrieved<br />
homologs have minimum 90% coverage with the query<br />
sequence. By using HHfilter, a post processing tool<br />
provided in the HHblits package, we built two different<br />
sets of MSAs by varying the maximum pairwise sequence<br />
identity threshold between the collected homologs in each<br />
MSA. The (ungapped) sequences in the MSAs were<br />
predicted without normalisation in order to preserve the<br />
differences within a protein family, and mapped back to<br />
the full (gapped) MSA.<br />
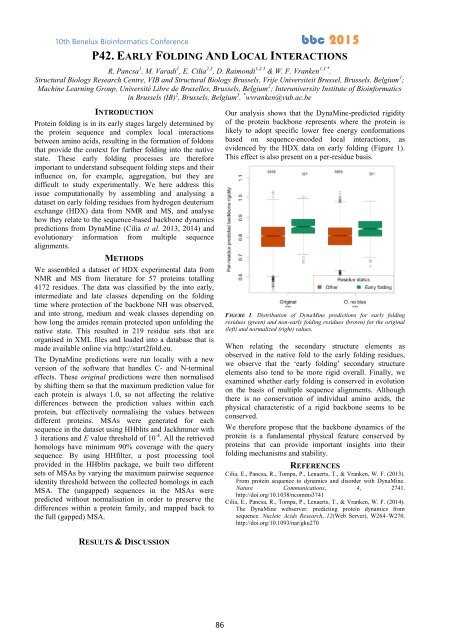
Our analysis shows that the DynaMine-predicted rigidity<br />
of the protein backbone represents where the protein is<br />
likely to adopt specific lower free energy conformations<br />
based on sequence-encoded local interactions, as<br />
evidenced by the HDX data on early folding (Figure 1).<br />
This effect is also present on a per-residue basis.<br />
FIGURE 1. Distribution of DynaMine predictions for early folding<br />
residues (green) and non-early folding residues (brown) for the original<br />
(left) and normalized (right) values.<br />
When relating the secondary structure elements as<br />
observed in the native fold to the early folding residues,<br />
we observe that the ‘early folding’ secondary structure<br />
elements also tend to be more rigid overall. Finally, we<br />
examined whether early folding is conserved in evolution<br />
on the basis of multiple sequence alignments. Although<br />
there is no conservation of individual amino acids, the<br />
physical characteristic of a rigid backbone seems to be<br />
conserved.<br />
We therefore propose that the backbone dynamics of the<br />
protein is a fundamental physical feature conserved by<br />
proteins that can provide important insights into their<br />
folding mechanisms and stability.<br />
REFERENCES<br />
Cilia, E., Pancsa, R., Tompa, P., Lenaerts, T., & Vranken, W. F. (2013).<br />
From protein sequence to dynamics and disorder with DynaMine.<br />
Nature Communications, 4, 2741.<br />
http://doi.org/10.1038/ncomms3741<br />
Cilia, E., Pancsa, R., Tompa, P., Lenaerts, T., & Vranken, W. F. (2014).<br />
The DynaMine webserver: predicting protein dynamics from<br />
sequence. Nucleic Acids Research, 12(Web Server), W264–W270.<br />
http://doi.org/10.1093/nar/gku270<br />
RESULTS & DISCUSSION<br />
86