

The Genom of Homo sapiens.pdf
The Genom of Homo sapiens.pdf
The Genom of Homo sapiens.pdf
- TAGS
- homo
- www.yumpu.com

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
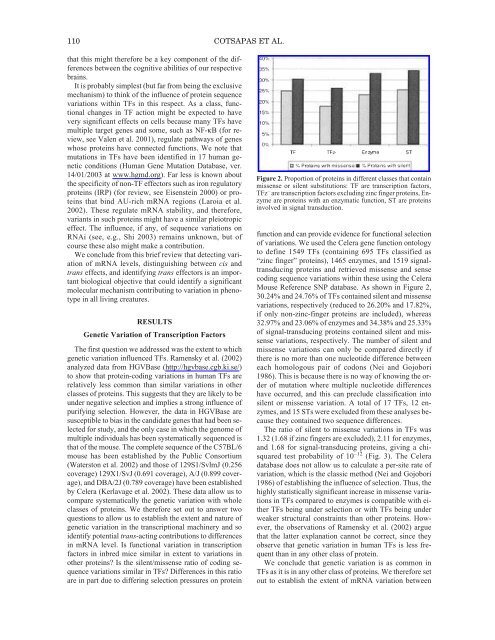
110 COTSAPAS ET AL.that this might therefore be a key component <strong>of</strong> the differencesbetween the cognitive abilities <strong>of</strong> our respectivebrains.It is probably simplest (but far from being the exclusivemechanism) to think <strong>of</strong> the influence <strong>of</strong> protein sequencevariations within TFs in this respect. As a class, functionalchanges in TF action might be expected to havevery significant effects on cells because many TFs havemultiple target genes and some, such as NF-κB (for review,see Valen et al. 2001), regulate pathways <strong>of</strong> geneswhose proteins have connected functions. We note thatmutations in TFs have been identified in 17 human geneticconditions (Human Gene Mutation Database, ver.14/01/2003 at www.hgmd.org). Far less is known aboutthe specificity <strong>of</strong> non-TF effectors such as iron regulatoryproteins (IRP) (for review, see Eisenstein 2000) or proteinsthat bind AU-rich mRNA regions (Laroia et al.2002). <strong>The</strong>se regulate mRNA stability, and therefore,variants in such proteins might have a similar pleiotropiceffect. <strong>The</strong> influence, if any, <strong>of</strong> sequence variations onRNAi (see, e.g., Shi 2003) remains unknown, but <strong>of</strong>course these also might make a contribution.We conclude from this brief review that detecting variation<strong>of</strong> mRNA levels, distinguishing between cis andtrans effects, and identifying trans effectors is an importantbiological objective that could identify a significantmolecular mechanism contributing to variation in phenotypein all living creatures.RESULTSGenetic Variation <strong>of</strong> Transcription Factors<strong>The</strong> first question we addressed was the extent to whichgenetic variation influenced TFs. Ramensky et al. (2002)analyzed data from HGVBase (http://hgvbase.cgb.ki.se/)to show that protein-coding variations in human TFs arerelatively less common than similar variations in otherclasses <strong>of</strong> proteins. This suggests that they are likely to beunder negative selection and implies a strong influence <strong>of</strong>purifying selection. However, the data in HGVBase aresusceptible to bias in the candidate genes that had been selectedfor study, and the only case in which the genome <strong>of</strong>multiple individuals has been systematically sequenced isthat <strong>of</strong> the mouse. <strong>The</strong> complete sequence <strong>of</strong> the C57BL/6mouse has been established by the Public Consortium(Waterston et al. 2002) and those <strong>of</strong> 129S1/SvlmJ (0.256coverage) 129X1/SvJ (0.691 coverage), A/J (0.899 coverage),and DBA/2J (0.789 coverage) have been establishedby Celera (Kerlavage et al. 2002). <strong>The</strong>se data allow us tocompare systematically the genetic variation with wholeclasses <strong>of</strong> proteins. We therefore set out to answer twoquestions to allow us to establish the extent and nature <strong>of</strong>genetic variation in the transcriptional machinery and soidentify potential trans-acting contributions to differencesin mRNA level. Is functional variation in transcriptionfactors in inbred mice similar in extent to variations inother proteins? Is the silent/missense ratio <strong>of</strong> coding sequencevariations similar in TFs? Differences in this ratioare in part due to differing selection pressures on proteinFigure 2. Proportion <strong>of</strong> proteins in different classes that containmissense or silent substitutions: TF are transcription factors,TFz – are transcription factors excluding zinc finger proteins, Enzymeare proteins with an enzymatic function, ST are proteinsinvolved in signal transduction.function and can provide evidence for functional selection<strong>of</strong> variations. We used the Celera gene function ontologyto define 1549 TFs (containing 695 TFs classified as“zinc finger” proteins), 1465 enzymes, and 1519 signaltransducingproteins and retrieved missense and sensecoding sequence variations within these using the CeleraMouse Reference SNP database. As shown in Figure 2,30.24% and 24.76% <strong>of</strong> TFs contained silent and missensevariations, respectively (reduced to 26.20% and 17.82%,if only non-zinc-finger proteins are included), whereas32.97% and 23.06% <strong>of</strong> enzymes and 34.38% and 25.33%<strong>of</strong> signal-transducing proteins contained silent and missensevariations, respectively. <strong>The</strong> number <strong>of</strong> silent andmissense variations can only be compared directly ifthere is no more than one nucleotide difference betweeneach homologous pair <strong>of</strong> codons (Nei and Gojobori1986). This is because there is no way <strong>of</strong> knowing the order<strong>of</strong> mutation where multiple nucleotide differenceshave occurred, and this can preclude classification intosilent or missense variation. A total <strong>of</strong> 17 TFs, 12 enzymes,and 15 STs were excluded from these analyses becausethey contained two sequence differences.<strong>The</strong> ratio <strong>of</strong> silent to missense variations in TFs was1.32 (1.68 if zinc fingers are excluded), 2.11 for enzymes,and 1.68 for signal-transducing proteins, giving a chisquaredtest probability <strong>of</strong> 10 –12 (Fig. 3). <strong>The</strong> Celeradatabase does not allow us to calculate a per-site rate <strong>of</strong>variation, which is the classic method (Nei and Gojobori1986) <strong>of</strong> establishing the influence <strong>of</strong> selection. Thus, thehighly statistically significant increase in missense variationsin TFs compared to enzymes is compatible with eitherTFs being under selection or with TFs being underweaker structural constraints than other proteins. However,the observations <strong>of</strong> Ramensky et al. (2002) arguethat the latter explanation cannot be correct, since theyobserve that genetic variation in human TFs is less frequentthan in any other class <strong>of</strong> protein.We conclude that genetic variation is as common inTFs as it is in any other class <strong>of</strong> proteins. We therefore setout to establish the extent <strong>of</strong> mRNA variation between













