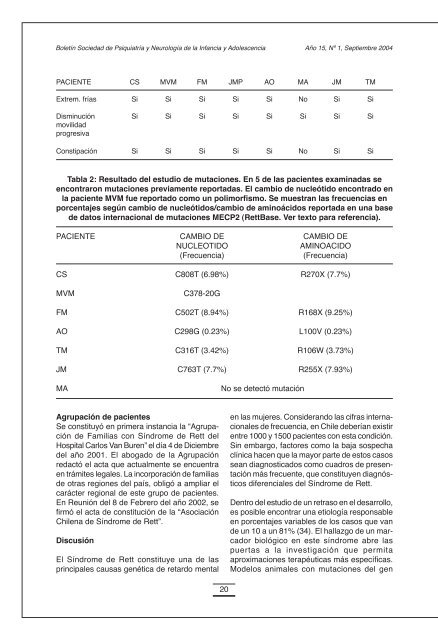
Boletín Sociedad <strong>de</strong> Psiquiatría y Neurología <strong>de</strong> <strong>la</strong> Infancia y Adolescencia Año 15, Nº 1, Septiembre 2004PACIENTE CS MVM FM JMP AO MA JM TMExtrem. frías Si Si Si Si Si No Si SiDisminución Si Si Si Si Si Si Si SimovilidadprogresivaConstipación Si Si Si Si Si No Si SiTab<strong>la</strong> 2: Resultado <strong>de</strong>l estudio <strong>de</strong> mutaciones. En 5 <strong>de</strong> <strong>la</strong>s pacientes examinadas seencontraron mutaciones previamente reportadas. El cambio <strong>de</strong> nucleótido encontrado en<strong>la</strong> paciente MVM fue reportado como un polimorfismo. Se muestran <strong>la</strong>s frecuencias enporcentajes según cambio <strong>de</strong> nucleótidos/cambio <strong>de</strong> aminoácidos reportada en una base<strong>de</strong> datos internacional <strong>de</strong> mutaciones MECP2 (RettBase. Ver texto para referencia).PACIENTE CAMBIO DE CAMBIO DENUCLEOTIDOAMINOACIDO(Frecuencia)(Frecuencia)CS C808T (6.98%) R270X (7.7%)MVMC378-20GFM C502T (8.94%) R168X (9.25%)AO C298G (0.23%) L100V (0.23%)TM C316T (3.42%) R106W (3.73%)JM C763T (7.7%) R255X (7.93%)MA No se <strong>de</strong>tectó mutaciónAgrupación <strong>de</strong> pacientesSe constituyó en primera instancia <strong>la</strong> “Agrupación<strong>de</strong> Familias con Síndrome <strong>de</strong> Rett <strong>de</strong>lHospital Carlos Van Buren” el día 4 <strong>de</strong> Diciembre<strong>de</strong>l año 2001. El abogado <strong>de</strong> <strong>la</strong> Agrupaciónredactó el acta que actualmente se encuentraen trámites legales. La incorporación <strong>de</strong> familias<strong>de</strong> otras regiones <strong>de</strong>l país, obligó a ampliar elcarácter regional <strong>de</strong> este grupo <strong>de</strong> pacientes.En Reunión <strong>de</strong>l 8 <strong>de</strong> Febrero <strong>de</strong>l año 2002, sefirmó el acta <strong>de</strong> constitución <strong>de</strong> <strong>la</strong> “AsociaciónChilena <strong>de</strong> Síndrome <strong>de</strong> Rett”.DiscusiónEl Síndrome <strong>de</strong> Rett constituye una <strong>de</strong> <strong>la</strong>sprincipales causas genética <strong>de</strong> retardo mentalen <strong>la</strong>s mujeres. Consi<strong>de</strong>rando <strong>la</strong>s cifras internacionales<strong>de</strong> frecuencia, en Chile <strong>de</strong>berían existirentre 1000 y 1500 pacientes con esta condición.Sin embargo, factores como <strong>la</strong> baja sospechaclínica hacen que <strong>la</strong> mayor parte <strong>de</strong> estos casossean diagnosticados como cuadros <strong>de</strong> presentaciónmás frecuente, que constituyen diagnósticosdiferenciales <strong>de</strong>l Síndrome <strong>de</strong> Rett.Dentro <strong>de</strong>l estudio <strong>de</strong> un retraso en el <strong>de</strong>sarrollo,es posible encontrar una etiología responsableen porcentajes variables <strong>de</strong> los casos que van<strong>de</strong> un 10 a un 81% (34). El hal<strong>la</strong>zgo <strong>de</strong> un marcadorbiológico en este síndrome abre <strong>la</strong>spuertas a <strong>la</strong> investigación que permitaaproximaciones terapéuticas más específicas.Mo<strong>de</strong>los animales con mutaciones <strong>de</strong>l gen20
Síndrome <strong>de</strong> RettJuan Francisco CabelloMECP2 han permitido explorar <strong>la</strong>s alteracionesen el <strong>de</strong>sarrollo <strong>de</strong>l sistema nervioso central quellevan a <strong>la</strong>s manifestaciones clínicas propias <strong>de</strong>lsíndrome y permiten postu<strong>la</strong>r alternativasterapéuticas como <strong>la</strong> terapia génica.Si consi<strong>de</strong>ramos pacientes con Síndrome <strong>de</strong>Rett típico o clásico a <strong>la</strong>s pacientes que cumplentodos los criterios diagnósticos mayoresestablecidos en nuestra ficha <strong>de</strong> inscripción, <strong>la</strong>corre<strong>la</strong>ción <strong>de</strong> <strong>la</strong> clínica presentada con elhal<strong>la</strong>zgo <strong>de</strong> mutaciones en el gen MECP2 fue<strong>de</strong> un 80% (4 <strong>de</strong> 5 pacientes) en este grupo <strong>de</strong>pacientes. Las mutaciones encontradas en 5<strong>de</strong> <strong>la</strong>s pacientes <strong>de</strong> esta serie clínica han sidopreviamente reportadas en <strong>la</strong> literatura. Losdatos <strong>de</strong> frecuencia obtenidos <strong>de</strong>s<strong>de</strong> una base<strong>de</strong> datos internacional <strong>de</strong> mutaciones MECP2(35), mostraron que <strong>la</strong> paciente FM tenía unamutación que ha sido encontrada en cerca <strong>de</strong>l9% <strong>de</strong> los casos reportados a esta base <strong>de</strong>datos, mientras que AO portaba <strong>la</strong> mutación <strong>de</strong>más rara ocurrencia <strong>de</strong>ntro <strong>de</strong>l grupo estudiado.Es importante <strong>de</strong>stacar <strong>la</strong> co<strong>la</strong>boración<strong>de</strong>sinteresada que el Instituto <strong>de</strong> GenéticaHumana <strong>de</strong> <strong>la</strong> Universidad <strong>de</strong> Goettingen haprestado a esta Agrupación <strong>de</strong> pacientes. Deacuerdo a lo publicado en literatura científicanacional, estas serían <strong>la</strong>s primeras pacienteschilenas con Síndrome <strong>de</strong> Rett a quienes seles ha practicado el estudio molecu<strong>la</strong>r paramutaciones <strong>de</strong> MECP2. En <strong>la</strong> paciente MVM,el hal<strong>la</strong>zgo <strong>de</strong> un polimorfismo motivó elp<strong>la</strong>nteamiento <strong>de</strong> que esta pudiera tratarse <strong>de</strong><strong>la</strong> primera oportunidad en que este cambio <strong>de</strong>nucleótidos fuera reportada, mas aun consi<strong>de</strong>randoel fenotipo clásico <strong>de</strong> presentación. Lapaciente MA no cumple criterios diagnósticosmayores y son pocos los criterios <strong>de</strong> soporteque apoyan el diagnóstico <strong>de</strong> Rett. Por esto, sele ha recomendado a <strong>la</strong> familia <strong>de</strong> MA ahondaren el estudio <strong>de</strong> algunos diagnósticos diferenciales.El retraso promedio en el diagnóstico <strong>de</strong> más<strong>de</strong> 17 meses <strong>de</strong>s<strong>de</strong> <strong>la</strong> aparición <strong>de</strong> los primerossíntomas pue<strong>de</strong> explicarse por <strong>la</strong> poca especificidad<strong>de</strong> los hal<strong>la</strong>zgos tempranos, y el <strong>de</strong>sconocimiento<strong>de</strong> <strong>la</strong> comunidad médica <strong>de</strong> este tipo<strong>de</strong> patologías.Luego <strong>de</strong> <strong>la</strong> formación <strong>de</strong> <strong>la</strong> “Agrupación <strong>de</strong>Familias con Síndrome <strong>de</strong> Rett <strong>de</strong>l HospitalCarlos Van Buren”, y <strong>de</strong>bido a <strong>la</strong> llegada <strong>de</strong>pacientes <strong>de</strong>s<strong>de</strong> otras regiones <strong>de</strong> Chile, <strong>de</strong>biócambiarse el nombre a “Asociación Chilena <strong>de</strong>Síndrome <strong>de</strong> Rett”, constituyéndose en <strong>la</strong>agrupación <strong>de</strong> pacientes con este síndrome conun mayor número <strong>de</strong> asociados en el país. Lacomprensión por parte <strong>de</strong> médicos <strong>de</strong> diferentesHospitales a lo <strong>la</strong>rgo <strong>de</strong>l país <strong>de</strong> <strong>la</strong> utilidad <strong>de</strong>incorporar a <strong>la</strong> Agrupación a sus pacientes,permitió que esta iniciativa local se transformaraen un movimiento a nivel nacional. A <strong>la</strong> fecha<strong>de</strong> entrega <strong>de</strong> esta tesis, se encuentran oficialmenteregistradas 14 familias, y 2 familias másestán en trámites <strong>de</strong> afiliación. Uno <strong>de</strong> estospacientes es un paciente <strong>de</strong> sexo masculino conun fenotipo clásico <strong>de</strong> Síndrome <strong>de</strong> Rett y <strong>la</strong>concomitancia <strong>de</strong> un Síndrome <strong>de</strong> Klinefelter.Se ha enviado muestra para análisis molecu<strong>la</strong>r<strong>de</strong> este paciente, que aún tiene pendiente suresultado.Los padres han organizado reuniones en quehan asistido pacientes <strong>de</strong> Puerto Aysen,Santiago, Pucón, Vil<strong>la</strong> Alemana, Santiago, Viña<strong>de</strong>l Mar, Valparaíso y Los Angeles.Actualmente están tramitando su personalidadjurídica y piensan adjudicarse <strong>la</strong> representación<strong>de</strong> <strong>la</strong> IRSA en Chile.ConclusionesEl Síndrome <strong>de</strong> Rett es un importantediagnóstico diferencial en niñas con retardomental y/o autismo. También <strong>de</strong>be serconsi<strong>de</strong>rado en pacientes <strong>de</strong> sexo masculinocon retardo mental y alteraciones asociadas.El diagnóstico <strong>de</strong>l Síndrome <strong>de</strong> Rett es eminentementeclínico. El hal<strong>la</strong>zgo <strong>de</strong> mutaciones <strong>de</strong>lgen MECP2 no es un sinónimo <strong>de</strong> Síndrome<strong>de</strong> Rett, pero es <strong>de</strong> utilidad para establecer undiagnóstico etiológico <strong>de</strong>finitivo, así como para<strong>la</strong> realización <strong>de</strong> estudios orientados aestablecer re<strong>la</strong>ciones fenotipo-genotipo yeventualmente proponer alternativas terapéuticas.La formación <strong>de</strong> Agrupaciones <strong>de</strong> pacientes conenfermeda<strong>de</strong>s poco frecuentes está orientadaa mejorar <strong>la</strong> calidad <strong>de</strong> vida <strong>de</strong> sus asociados y21