

Materialforschung mit Positronen: Von der Doppler-Spektroskopie zur
Materialforschung mit Positronen: Von der Doppler-Spektroskopie zur
Materialforschung mit Positronen: Von der Doppler-Spektroskopie zur

Erfolgreiche ePaper selbst erstellen
Machen Sie aus Ihren PDF Publikationen ein blätterbares Flipbook mit unserer einzigartigen Google optimierten e-Paper Software.
atio to anealed Al<br />
1.10<br />
1.05<br />
1.00<br />
Al<br />
10 -20h<br />
20 -30h<br />
50 - 60 h<br />
70 -80h<br />
0.95<br />
56<br />
0 10 20<br />
p L<br />
[10 -3 m 0<br />
c]<br />
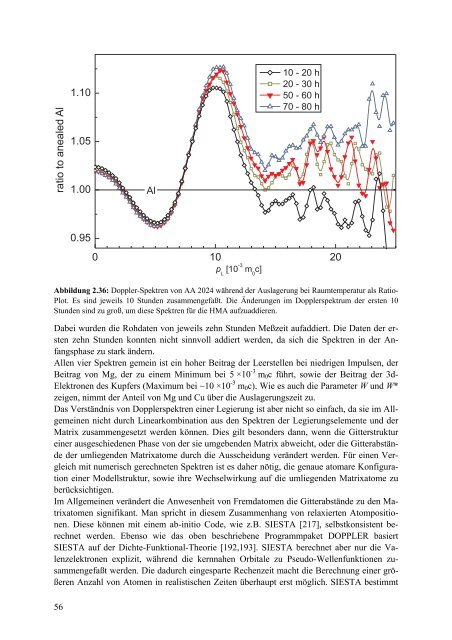
Abbildung 2.36: <strong>Doppler</strong>-Spektren von AA 2024 während <strong>der</strong> Auslagerung bei Raumtemperatur als Ratio-<br />
Plot. Es sind jeweils 10 Stunden zusammengefaßt. Die Än<strong>der</strong>ungen im <strong>Doppler</strong>spektrum <strong>der</strong> ersten 10<br />
Stunden sind zu groß, um diese Spektren für die HMA aufzuaddieren.<br />
Dabei wurden die Rohdaten von jeweils zehn Stunden Meßzeit aufaddiert. Die Daten <strong>der</strong> ersten<br />
zehn Stunden konnten nicht sinnvoll addiert werden, da sich die Spektren in <strong>der</strong> Anfangsphase<br />
zu stark än<strong>der</strong>n.<br />
Allen vier Spektren gemein ist ein hoher Beitrag <strong>der</strong> Leerstellen bei niedrigen Impulsen, <strong>der</strong><br />
Beitrag von Mg, <strong>der</strong> zu einem Minimum bei 5 ×10 -3 m 0 c führt, sowie <strong>der</strong> Beitrag <strong>der</strong> 3d-<br />
Elektronen des Kupfers (Maximum bei ~10 ×10 -3 m 0 c). Wie es auch die Parameter W und W*<br />
zeigen, nimmt <strong>der</strong> Anteil von Mg und Cu über die Auslagerungszeit zu.<br />
Das Verständnis von <strong>Doppler</strong>spektren einer Legierung ist aber nicht so einfach, da sie im Allgemeinen<br />
nicht durch Linearkombination aus den Spektren <strong>der</strong> Legierungselemente und <strong>der</strong><br />
Matrix zusammengesetzt werden können. Dies gilt beson<strong>der</strong>s dann, wenn die Gitterstruktur<br />
einer ausgeschiedenen Phase von <strong>der</strong> sie umgebenden Matrix abweicht, o<strong>der</strong> die Gitterabstände<br />
<strong>der</strong> umliegenden Matrixatome durch die Ausscheidung verän<strong>der</strong>t werden. Für einen Vergleich<br />
<strong>mit</strong> numerisch gerechneten Spektren ist es daher nötig, die genaue atomare Konfiguration<br />
einer Modellstruktur, sowie ihre Wechselwirkung auf die umliegenden Matrixatome zu<br />
berücksichtigen.<br />
Im Allgemeinen verän<strong>der</strong>t die Anwesenheit von Fremdatomen die Gitterabstände zu den Matrixatomen<br />
signifikant. Man spricht in diesem Zusammenhang von relaxierten Atompositionen.<br />
Diese können <strong>mit</strong> einem ab-initio Code, wie z.B. SIESTA [217], selbstkonsistent berechnet<br />
werden. Ebenso wie das oben beschriebene Programmpaket DOPPLER basiert<br />
SIESTA auf <strong>der</strong> Dichte-Funktional-Theorie [192,193]. SIESTA berechnet aber nur die Valenzelektronen<br />
explizit, während die kernnahen Orbitale zu Pseudo-Wellenfunktionen zusammengefaßt<br />
werden. Die dadurch eingesparte Rechenzeit macht die Berechnung einer größeren<br />
Anzahl von Atomen in realistischen Zeiten überhaupt erst möglich. SIESTA bestimmt




