- Page 1 and 2:
netLibrary - eBook Summary Universi
- Page 3 and 4:
Structure-based Drug Design 14 Stru
- Page 5 and 6:
Document p17, p9, and p7) and repli
- Page 7 and 8:
Document Figure 1 Stereo view of th
- Page 9 and 10:
Document Figure 2 Schematic represe
- Page 11 and 12:
Document TF—transframe, PR—prot
- Page 13 and 14:
Document compounds that utilize sim
- Page 15 and 16:
Document Page 11 retroviral substra
- Page 17 and 18:
Document Page 13 of the active-site
- Page 19 and 20:
Document Page 15 Thiazoles are less
- Page 21 and 22:
Document Page 17 the resulting chim
- Page 23 and 24:
Document http://legacy.netlibrary.c
- Page 25 and 26:
Document Figure 5 Stereo view AG134
- Page 27 and 28:
Document Page 23 potentially loweri
- Page 29 and 30:
Document http://legacy.netlibrary.c
- Page 31 and 32:
Document Page 27 volume of the acti
- Page 33 and 34:
Document Figure 7 Cartoon represent
- Page 35 and 36:
Document Figure 8 Cartoon represent
- Page 37 and 38:
Document Page 33 sustained in many
- Page 39 and 40:
Document 8. Kaplan AH, Zack JA, Kni
- Page 41 and 42:
Document 22. Krausslich HG, Ingraha
- Page 43 and 44:
Document 35. Kaldor SW, Hammond M,
- Page 45 and 46:
Document 47. Tummino PJ, Ferguson D
- Page 47 and 48:
Document 60. Condra JH, Schleif WA,
- Page 49 and 50:
Document 74. Reddy RM, Varney MD, K
- Page 51 and 52:
Document 2 Structural Studies of HI
- Page 53 and 54:
Document Figure 1 (a) Chemical stru
- Page 55 and 56:
Document dine (3TC), and 2',3'-dide
- Page 57 and 58:
Document Figure Continued Page 45 r
- Page 59 and 60:
Document Figure 2 Ribbon diagrams o
- Page 61 and 62:
Document Figure 3 Overall structure
- Page 63 and 64:
Document monophosphate analogs whic
- Page 65 and 66:
Document Analysis of various HIV-1
- Page 67 and 68:
Document Page 54 Table 2) [12,54].
- Page 69 and 70:
Document Page 56 It is also attract
- Page 71 and 72:
Document Table 3 HIV-1 RT Amino Aci
- Page 73 and 74:
Document ever, once an NNRTI is bou
- Page 75 and 76:
Document Page 61 now clear that the
- Page 77 and 78:
Document Page 63 most of the amino
- Page 79 and 80:
Document Page 65 cal properties of
- Page 81 and 82:
Document Page 67 Biochemical data s
- Page 83 and 84:
Document Page 69 determining the me
- Page 85 and 86:
Document Chris Tantillo. The work i
- Page 87 and 88:
Document Page 72 16. Goldman ME, Nu
- Page 89 and 90:
Document 30. Coffin JM. HIV populat
- Page 91 and 92:
Document 45. Majumadar C, Abbotts J
- Page 93 and 94:
Document reverse transcriptase inhi
- Page 95 and 96:
Document 75. Ring CS, Sun E, McKerr
- Page 97 and 98:
Document 89. Hughes SH, Arnold E, H
- Page 99 and 100:
Document 106. Cirino NM, Cameron CE
- Page 101 and 102:
Document 122. Marshall WS, Beaton G
- Page 103 and 104: Document dine) and didanosine (dide
- Page 105 and 106: Document 149. Craig JC, Duncan IB,
- Page 107 and 108: Document 163. Lacey SF, Larder BA.
- Page 109 and 110: Document Figure 1 Retroviral lifecy
- Page 111 and 112: Document Figure 2 In vivo reactions
- Page 113 and 114: Document Page 88 transfer (Figure 3
- Page 115 and 116: Document http://legacy.netlibrary.c
- Page 117 and 118: Document Page 92 The role of the N-
- Page 119 and 120: Document Figure 6 Molscript stereo
- Page 121 and 122: Document Page 96 region in the HIV-
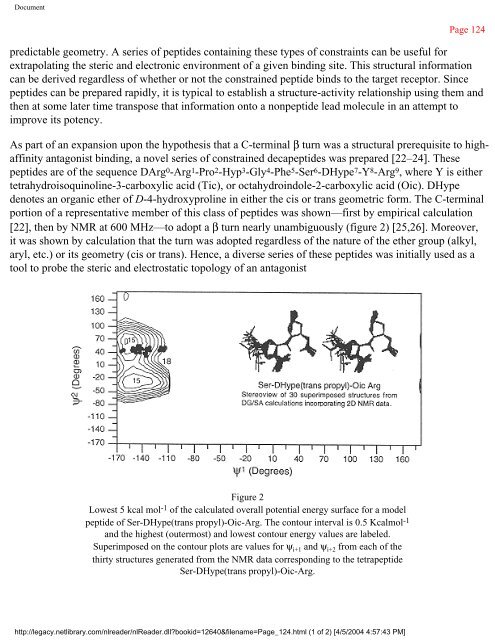
- Page 123 and 124: Document day junction resolving enz
- Page 125 and 126: Document -62° for HIV-1 integrase,
- Page 127 and 128: Document Page 101 crystallized unde
- Page 129 and 130: Document Figure 8 Molscript stereo
- Page 131 and 132: Document proposed mechanisms. For e
- Page 133 and 134: Document Page 106 on the same ring
- Page 135 and 136: Document Page 108 mulate during tre
- Page 137 and 138: Document when metals are bound. Fin
- Page 139 and 140: Document cubation of phosphotyrosin
- Page 141 and 142: Document 4. Vink C, Plasterk RHA. T
- Page 143 and 144: Document Page 115 21. Kulkosky J, J
- Page 145 and 146: Document 44. Cushman M, Sherman P.
- Page 147 and 148: Document 65. Gallay P, Swingler S,
- Page 149 and 150: Document 4 Bradykinin Receptor Anta
- Page 151 and 152: Document Page 121 Nearly all cells
- Page 153: Document Page 123 were poorly satis
- Page 157 and 158: Document Page 127 The des-Arg 9 for
- Page 159 and 160: Document Page 129 tolerated by the
- Page 161 and 162: Document receptor, it has not yet b
- Page 163 and 164: Document Page 133 This initial stag
- Page 165 and 166: Document Page 135 might be jointly
- Page 167 and 168: Document Page 137 observed for the
- Page 169 and 170: Document Figure 6 Rat and human B2
- Page 171 and 172: Document Page 141 receptor with int
- Page 173 and 174: Document http://legacy.netlibrary.c
- Page 175 and 176: Document Figure 9 Composition of te
- Page 177 and 178: Document http://legacy.netlibrary.c
- Page 179 and 180: Document 16. Martorana PA, Kettenba
- Page 181 and 182: Document 39. Mavunkel BJ, Lu Z, Kyl
- Page 183 and 184: http://legacy.netlibrary.com/reader
- Page 185 and 186: Document B. Pharmacology Figure 1 T
- Page 187 and 188: Document We determined the structur
- Page 189 and 190: Document Figure 3 Previously known
- Page 191 and 192: Document http://legacy.netlibrary.c
- Page 193 and 194: Document http://legacy.netlibrary.c
- Page 195 and 196: Document large solvent channels and
- Page 197 and 198: Document IV. Drug Design Progressio
- Page 199 and 200: Document position eight from formin
- Page 201 and 202: Document Table 1 Inhibition Data fo
- Page 203 and 204: Document Page 166 As predicated, th
- Page 205 and 206:
Document References 1. Parks RE Jr.
- Page 207 and 208:
Document 17. Ealick SE, Rule SA, Ca
- Page 209 and 210:
Document 6 Structural Implications
- Page 211 and 212:
Document Figure 1 A ribbon model of
- Page 213 and 214:
Document http://legacy.netlibrary.c
- Page 215 and 216:
Document Page 176 0.43 Å) as the c
- Page 217 and 218:
Document Page 178 studies show that
- Page 219 and 220:
Document Figure 5 (a) A cut away vi
- Page 221 and 222:
Document Page 182 (SAR) model. The
- Page 223 and 224:
Document IX. S2' Interactions Page
- Page 225 and 226:
Document Figure 6 (a) The pocket of
- Page 227 and 228:
Document 5. Willenbrock F, Murphy G
- Page 229 and 230:
Document 20. Murphy G, Docherty AJP
- Page 231 and 232:
Document Page 189 36. Bode W, Reine
- Page 233 and 234:
Document 7 Structure—Function Rel
- Page 235 and 236:
Document Figure 1 Reactions catalyz
- Page 237 and 238:
Document Figure 2 Structure of lico
- Page 239 and 240:
Document Figure 3 (Continued) Page
- Page 241 and 242:
Document Important for the validity
- Page 243 and 244:
Document Figure 4 Amino acids impor
- Page 245 and 246:
Document III. Results and Discussio
- Page 247 and 248:
Document Page 202 269, and valine-2
- Page 249 and 250:
Document Figure 6 Structure of α h
- Page 251 and 252:
Document Page 205 enzyme and of phe
- Page 253 and 254:
Document Page 207 sure and the acti
- Page 255 and 256:
Document 13. Wilson RC, Krozowski Z
- Page 257 and 258:
Document 29. Baker ME. Genealogy of
- Page 259 and 260:
Document 46. Ribas dePoplana L, Fot
- Page 261 and 262:
http://legacy.netlibrary.com/reader
- Page 263 and 264:
Document Page 214 viruses. Specific
- Page 265 and 266:
Document Figure 1 (Continued) this
- Page 267 and 268:
Document Page 218 The catalytic loo
- Page 269 and 270:
Document Page 220 conserved substra
- Page 271 and 272:
Document Figure 3 (a) Ternary compl
- Page 273 and 274:
Document Figure 5 (a) Staurosporine
- Page 275 and 276:
Document protein kinase core is ess
- Page 277 and 278:
Document 10. Knighton DR, Zheng J-H
- Page 279 and 280:
Document 24. Madhusudan Xuong N-H,
- Page 281 and 282:
Document 9 Structural Studies of Al
- Page 283 and 284:
Document diverse ARIs have been sho
- Page 285 and 286:
Document Figure 3 C α trace of the
- Page 287 and 288:
Document Figure 4 Surface represent
- Page 289 and 290:
Document Figure 5 Schematic represe
- Page 291 and 292:
Document Figure 7 Stereo of zopolre
- Page 293 and 294:
Document B. Structures Figure 8 Seq
- Page 295 and 296:
Document This method was used to sc
- Page 297 and 298:
Document 2. Lee AYW, Chung SK, Chun
- Page 299 and 300:
Document 18. Wilson DK, Tarle I, Pe
- Page 301 and 302:
Document 35. Pailhoux EA, Martinez
- Page 303 and 304:
Document 10 Structure-Based Design
- Page 305 and 306:
Document Figure 2 Schematic represe
- Page 307 and 308:
Document Figure 4 Schematic represe
- Page 309 and 310:
Document Figure 6 Schematic represe
- Page 311 and 312:
Document Cyclotheonamide A (CtA), a
- Page 313 and 314:
Document Page 256 The discovery pro
- Page 315 and 316:
Document http://legacy.netlibrary.c
- Page 317 and 318:
Document et al. [21]). Among these
- Page 319 and 320:
Document balance its pro- and antic
- Page 321 and 322:
Document Page 263 15. Tabernero L,
- Page 323 and 324:
Document 31. Kettner C, Shaw E. D-P
- Page 325 and 326:
Document Figure 1 The coagulation c
- Page 327 and 328:
Document Figure 2 Factor Xa structu
- Page 329 and 330:
Document Table 2 Naturally Occurrin
- Page 331 and 332:
Document Table 3 Active Site Sequen
- Page 333 and 334:
Document Figure 5 Predicted seconda
- Page 335 and 336:
Document Lys in the P1 position is
- Page 337 and 338:
Document Page 276 In both cases the
- Page 339 and 340:
Document Page 278 The n=3 chain len
- Page 341 and 342:
Document Figure 10 Proposed model o
- Page 343 and 344:
Document Figure 11 dArg-ATS 32-38 m
- Page 345 and 346:
Document Figure 12 Modeled fit of S
- Page 347 and 348:
Document Page 285 number of x-ray s
- Page 349 and 350:
Document http://legacy.netlibrary.c
- Page 351 and 352:
Document Page 288 Additionally, the
- Page 353 and 354:
Document Page 290 5. Kaiser B, Haup
- Page 355 and 356:
Document Page 291 21. Davie EW, Fuj
- Page 357 and 358:
Document 36. Leytus SP, Chung DW, K
- Page 359 and 360:
Document 54. Broze Jr GJ, Girard TJ
- Page 361 and 362:
Document Page 294 70. Seligmann B,
- Page 363 and 364:
Document 12 Polypeptide Modulators
- Page 365 and 366:
Document Page 297 whereas, calcium
- Page 367 and 368:
Document Figure 2 Amino acid sequen
- Page 369 and 370:
Document Page 301 domains contribut
- Page 371 and 372:
Document Figure 5 Stereo views of 2
- Page 373 and 374:
Document A. Chemical Modification P
- Page 375 and 376:
Document Page 307 (the exception is
- Page 377 and 378:
Document Page 309 site 3 on the sod
- Page 379 and 380:
Document http://legacy.netlibrary.c
- Page 381 and 382:
Document Page 312 In peptide—prot
- Page 383 and 384:
Document Page 314 anemone toxins an
- Page 385 and 386:
Document 5. Packer M, Gheorghiade M
- Page 387 and 388:
Document 25. Malpezzi ELA, De Freit
- Page 389 and 390:
Document 42. Catterall WA, Beress L
- Page 391 and 392:
Document 60. Pennington MW, Zadenbe
- Page 393 and 394:
Document 76. Gould AR, Mabbutt BC,
- Page 395 and 396:
Document 13 Rational Design of Reni
- Page 397 and 398:
Document Page 323 been suggested th
- Page 399 and 400:
Document Figure 2 A schematic diagr
- Page 401 and 402:
Document Page 327 this analog inter
- Page 403 and 404:
Document Page 329 binding involves
- Page 405 and 406:
Document plasma. This may arise fro
- Page 407 and 408:
Document C. Specificity Figure 4 Th
- Page 409 and 410:
Document Figure 5 The S 3 specifici
- Page 411 and 412:
Document IV. Rational Drug Design F
- Page 413 and 414:
Document 2. Blundell TL, Cooper J,
- Page 415 and 416:
Document 17. Szelke M. Chemistry of
- Page 417 and 418:
Document 34. Rosenberg SH, Plattner
- Page 419 and 420:
Document 48. Powers JC, Harley AD,
- Page 421 and 422:
Document Page 343 14 Structural Asp
- Page 423 and 424:
Document Figure 2 The reaction cata
- Page 425 and 426:
Document Figure 4 Amino acid sequen
- Page 427 and 428:
Document Figure 5 Schematic stereo
- Page 429 and 430:
Document Figure 8 The catalytic mac
- Page 431 and 432:
Document Figure 10 Structures of so
- Page 433 and 434:
Document From quantitative structur
- Page 435 and 436:
Document Page 355 with the tryptoph
- Page 437 and 438:
Document Figure 14 The energy profi
- Page 439 and 440:
Document Page 359 easily reach the
- Page 441 and 442:
Document Page 361 2. Guldberg H, Ma
- Page 443 and 444:
Document Page 362 19. Lotta T, Vidg
- Page 445 and 446:
Document 35. Davis TL, Roznoski M,
- Page 447 and 448:
http://legacy.netlibrary.com/reader
- Page 449 and 450:
Document Figure 1 Available drugs f
- Page 451 and 452:
Document Table 1 Trypanosomal Targe
- Page 453 and 454:
Document Table 2 Three-Dimensional
- Page 455 and 456:
Document Figure 2 Glycolysis in blo
- Page 457 and 458:
Document II. Three Glycolytic Enzym
- Page 459 and 460:
Document Figure 4 Stereoview of sup
- Page 461 and 462:
Document Figure 6 Stereoview of NAD
- Page 463 and 464:
Document Page 376 was recently solv
- Page 465 and 466:
Document Because there are no known
- Page 467 and 468:
Document Page 379 For trypanosomal
- Page 469 and 470:
Document http://legacy.netlibrary.c
- Page 471 and 472:
Document Because there are tens of
- Page 473 and 474:
Document 5-Methoxytryptamine 19.0 9
- Page 475 and 476:
Document Figure 11 Two-dimensional
- Page 477 and 478:
Document Figure 12 Predicted bindin
- Page 479 and 480:
Document Page 388 nosine proved to
- Page 481 and 482:
Document 15. Carter NS, Fairlamb AH
- Page 483 and 484:
Document 30. Kim H, Feil I, Verlind
- Page 485 and 486:
Document 49. Michels PAM, Hannaert
- Page 487 and 488:
Document 65. João HC, Williams RJP
- Page 489 and 490:
Document 84. Verlinde CLMJ, Hol WGJ
- Page 491 and 492:
Document 16 Progress in the Design
- Page 493 and 494:
Document Table 1 Known Three-Dimens
- Page 495 and 496:
Document Page 398 with growth hormo
- Page 497 and 498:
Document All effects of the IL-1 fa
- Page 499 and 500:
Document has a single membrane-span
- Page 501 and 502:
Document Figure 2 Structural alignm
- Page 503 and 504:
Document Page 405 strands constitut
- Page 505 and 506:
Document Figure 4 (a) Stereo diagra
- Page 507 and 508:
Document Figure 6 (a) Stereo diagra
- Page 509 and 510:
Document Figure 7 (a) Superposition
- Page 511 and 512:
Document Page 412 ture is unlike ot
- Page 513 and 514:
Document Figure 10 Schematic diagra
- Page 515 and 516:
Document Page 416 positions of the
- Page 517 and 518:
Document Figure 11 Functional resid
- Page 519 and 520:
Document Figure 13 The identified p
- Page 521 and 522:
Document B. Interleukin-1 Receptor
- Page 523 and 524:
Document Page 423 American Home Pro
- Page 525 and 526:
Document Page 425 Antinflammatory D
- Page 527 and 528:
Document Page 427 These aromatic di
- Page 529 and 530:
Document 5. Dinarello CA. On the Bi
- Page 531 and 532:
Document 21. Seckinger P, Lowenthal
- Page 533 and 534:
Document 39. Saurat JH, Schfferli J
- Page 535 and 536:
Document receptor antagonist into a
- Page 537 and 538:
Document 74. Bender PE, Lee JC., ed
- Page 539 and 540:
Document 102. Machin PJ, Osbond JM,
- Page 541 and 542:
Document 17 Structure and Functiona
- Page 543 and 544:
Document Table 1 Approval Indicatio
- Page 545 and 546:
Document Page 438 proteins. One pot
- Page 547 and 548:
Document II. Type IFNs A great deal
- Page 549 and 550:
Document Page 441 sequence of the m
- Page 551 and 552:
Document Page 443 that allowed for
- Page 553 and 554:
Document Figure 4 Stereo view of IF
- Page 555 and 556:
Document Figure 5 Velcro-key model
- Page 557 and 558:
Document Figure 6 Proposed receptor
- Page 559 and 560:
Document Page 451 with the cytoplas
- Page 561 and 562:
Document for directed subcellular t
- Page 563 and 564:
Document 13. Stewart HJ, McCann SHE
- Page 565 and 566:
Document 33. Szente BE, Johnson HM.
- Page 567 and 568:
Document 47. Igarashi K, Garotta G,
- Page 569 and 570:
http://legacy.netlibrary.com/reader
- Page 571 and 572:
Document Figure 1 A schematic diagr
- Page 573 and 574:
Document Page 462 strains of influe
- Page 575 and 576:
Document Page 464 could not again b
- Page 577 and 578:
Document Figure 3 (a) A MOLSCRIPT [
- Page 579 and 580:
Document B. Protein Structure Page
- Page 581 and 582:
Document Figure 5 (a) Stereo image
- Page 583 and 584:
Document Page 471 molecules in the
- Page 585 and 586:
Document hydroxy, and 6 Neu5Ac2en -
- Page 587 and 588:
Document Figure 8 Stereo image of t
- Page 589 and 590:
Document Page 476 nM, respectively.
- Page 591 and 592:
Document Page 478 lished—was asso
- Page 593 and 594:
Document VI. Conclusion Page 480 It
- Page 595 and 596:
Document 21. Both GW, Sleigh MJ, Co
- Page 597 and 598:
Document 40. Drzenick R, Frank H, R
- Page 599 and 600:
Document 45. Varghese JN, Laver WG,
- Page 601 and 602:
Document 64. Ward CW, Elleman TC, A
- Page 603 and 604:
Document 80. Suzuki Y, Sato K, Kiso
- Page 605 and 606:
Document Page 486 95. Ryan DM, Tice
- Page 607 and 608:
Document Page 488 against most of t
- Page 609 and 610:
Document http://legacy.netlibrary.c
- Page 611 and 612:
Document A. The Canyon Page 491 The
- Page 613 and 614:
Document Subsequent studies have sh
- Page 615 and 616:
Document Page 495 of the RNA and pr
- Page 617 and 618:
Document Figure 4 A ribbon diagram
- Page 619 and 620:
Document Figure 5 Some compounds wh
- Page 621 and 622:
Document Figure 7 HRV14 VP1 hydroph
- Page 623 and 624:
Document C. Structure-Activity Rela
- Page 625 and 626:
Document http://legacy.netlibrary.c
- Page 627 and 628:
Document http://legacy.netlibrary.c
- Page 629 and 630:
Document Hydrophobicity Requirement
- Page 631 and 632:
Document Figure 9 A solvent-accessi
- Page 633 and 634:
Document accommodate a variety of d
- Page 635 and 636:
Document Figure 10 Possible hydroge
- Page 637 and 638:
Document Page 514 sis. Large number
- Page 639 and 640:
Document conformational transition
- Page 641 and 642:
Document Page 518 The differences b
- Page 643 and 644:
Document 10. Mast EE, Harmon MW, Gr
- Page 645 and 646:
Document 25. Arnold E, Rossmann MG.
- Page 647 and 648:
Document 44. Argos P, Rossmann MG,
- Page 649 and 650:
Document 63. Rozhon E, Cox S, Buont
- Page 651 and 652:
Document 81. Diana GD, McKinlay MA,
- Page 653 and 654:
Document 20 The Integration of Stru
- Page 655 and 656:
Document Page 527 function. Iterati
- Page 657 and 658:
Document Figure 3 Generation of com
- Page 659 and 660:
Document Page 530 screen. For examp
- Page 661 and 662:
Document ciency of drug discovery (
- Page 663 and 664:
Document Page 533 the reactions are
- Page 665 and 666:
Document Page 535 by factoring in a
- Page 667 and 668:
Document Page 537 ries, holds the p
- Page 669 and 670:
Document 17. Zuckermann RN, Martin
- Page 671 and 672:
Document 34. Fei Y-J, Kanai Y, Nuss
- Page 673 and 674:
Document 21 Structure-Based Combina
- Page 675 and 676:
Document Figure 1 Schematic represe
- Page 677 and 678:
Document Page 545 protein binding s
- Page 679 and 680:
Document Figure 2 Stereo view of th
- Page 681 and 682:
Document Figure 3 (a) Stereo view o
- Page 683 and 684:
Document Figure 5 Stereo view of th
- Page 685 and 686:
Document Lists of Bonding Fragment
- Page 687 and 688:
Document Figure 8 Minimized structu
- Page 689 and 690:
Document Page 555 N-acylpyrrolidine
- Page 691 and 692:
Document 10. Bobbyer DNA, Goodford
- Page 693 and 694:
Document structure. 2. Ligand probe
- Page 695 and 696:
Document 30. O'Shea EK, Rutkowski R
- Page 697 and 698:
Document 22 Peptidomimetic and Nonp
- Page 699 and 700:
Document Figure 1 Examples of nativ
- Page 701 and 702:
Document B. Peptidomimetic Drugs: C
- Page 703 and 704:
Document Figure 4 Backbone amide bo
- Page 705 and 706:
Document 12640-0567a.gif Figure 6 C
- Page 707 and 708:
Document Page 569 molecule. A nonco
- Page 709 and 710:
Document cases, a pharmacophore mod
- Page 711 and 712:
Document Figure 9 Peptide scaffold-
- Page 713 and 714:
Document Figure 11 Protease-targete
- Page 715 and 716:
Document Figure 12 Peptide scaffold
- Page 717 and 718:
Document Figure 13 Peptide scaffold
- Page 719 and 720:
Document Figure 14 Signal-transduct
- Page 721 and 722:
Document Figure 15 Peptide scaffold
- Page 723 and 724:
Document Figure 16 Peptide scaffold
- Page 725 and 726:
Document Figure 17 Nonpeptide drug
- Page 727 and 728:
Document http://legacy.netlibrary.c
- Page 729 and 730:
Document http://legacy.netlibrary.c
- Page 731 and 732:
Document Page 591 often suggest tha
- Page 733 and 734:
Document Page 593 versus antagonist
- Page 735 and 736:
Document Page 595 the MC1 receptor
- Page 737 and 738:
Document http://legacy.netlibrary.c
- Page 739 and 740:
Document Figure 22 Protease 3D stru
- Page 741 and 742:
Document Page 601 inhibitors 100 [1
- Page 743 and 744:
Document Page 603 hydroxymethyl sub
- Page 745 and 746:
Document Page 605 For other serinyl
- Page 747 and 748:
Document Page 607 scopic studies pr
- Page 749 and 750:
Document Page 609 (e.g., thermolysi
- Page 751 and 752:
Document Figure 28 Protease 3D stru
- Page 753 and 754:
Document Figure 29 Protease 3D stru
- Page 755 and 756:
Document (Ser/Thr) peptide 2.9 Å 2
- Page 757 and 758:
Document http://legacy.netlibrary.c
- Page 759 and 760:
Document http://legacy.netlibrary.c
- Page 761 and 762:
Document Page 617 Val (131) showed
- Page 763 and 764:
Document Figure 32 PTP and PTB 3D s
- Page 765 and 766:
Document Acknowledgments Page 620 I
- Page 767 and 768:
Document 18. Sawyer TK. In: Peptide
- Page 769 and 770:
Document Page 622 Fukuroda T, Fukam
- Page 771 and 772:
Document Page 623 53. Bird J, Harpe
- Page 773 and 774:
Document 78. Rodriguez M, Crosby R,
- Page 775 and 776:
Document MA, Welch KM, Hallak H, Ta
- Page 777 and 778:
Document Page 626 SJ, Ogden RC, Red
- Page 779 and 780:
Document 97; (e) Fujii I, Nakamura
- Page 781 and 782:
Document Med Biol 1991; 306:9-21; (
- Page 783 and 784:
Document Page 629 ML, Clare M, Decr
- Page 785 and 786:
Document Page 630 177. (a) Bode W,
- Page 787 and 788:
Document Page 631 197. Musil D, Zuc
- Page 789 and 790:
Document son AH, Drummond AH, Huxle
- Page 791 and 792:
Document 236. (a) Marshall MS. TIBS
- Page 793 and 794:
Document http://legacy.netlibrary.c
- Page 795 and 796:
Document 274. (a) Kavanaugh WM, Wil
- Page 797 and 798:
Document replacements, 563-565 1-am
- Page 799 and 800:
Document inhibitor binding, 323, 33
- Page 801 and 802:
Document site, 52, 55, 61 triad, 24
- Page 803 and 804:
Document factors, 247 Combination t
- Page 805 and 806:
Document Cyclotheonamide A (CtA), 2
- Page 807 and 808:
Document D ddI, 152 tumour necrosis
- Page 809 and 810:
Document antistasin peptides, 281 c
- Page 811 and 812:
Document fragment-based programs, 5
- Page 813 and 814:
Document [Human immunodeficiency vi
- Page 815 and 816:
Document overview, 103-104, 108-109
- Page 817 and 818:
Document [Inhibitors] 2-((3,4-dihyd
- Page 819 and 820:
Document antagonistic activity, 416
- Page 821 and 822:
Document [Interleukin-1] homology w
- Page 823 and 824:
Document Kininogen, 119 high molecu
- Page 825 and 826:
Document Matrix-metalloproteinase (
- Page 827 and 828:
Document RT inhibitor, 41, 56 Nonpe
- Page 829 and 830:
Document Phosphoglycerate kinase (P
- Page 831:
Document [Protease targets] serinyl