

Alberto Risueño Pérez - Gredos - Universidad de Salamanca
Alberto Risueño Pérez - Gredos - Universidad de Salamanca
Alberto Risueño Pérez - Gredos - Universidad de Salamanca

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Tesis Doctoral<br />
inclusión entre 0 y 1 calculado mediante una aproximación bayesiana. La diferencia entre los<br />
valores <strong>de</strong> inclusión entre los dos tejidos es lo que da el nivel <strong>de</strong> confianza (score) <strong>de</strong> estar<br />
realmente ante un evento <strong>de</strong> splicing alternativo. Finalmente para nuestro estudio en esta<br />
Tesis Doctoral será consi<strong>de</strong>rado únicamente el conjunto <strong>de</strong> tejidos comunes a los dos sets <strong>de</strong><br />
datos citados: el set <strong>de</strong> microarrays <strong>de</strong> Affymetrix y el trabajo <strong>de</strong> Wang et al. (ver tabla 3.1).<br />
El conjunto <strong>de</strong> tejidos comunes a ambos grupos correspon<strong>de</strong> con 6 tejidos distintos: mama,<br />
cerebelo, corazón, hígado, músculo y testículo. La combinación <strong>de</strong> estos 6 tejidos cuando se<br />
comparan <strong>de</strong> 2 en 2 proporciona un total <strong>de</strong> 15 pares distintos. El número total <strong>de</strong> pares (gen ::<br />
combinación <strong>de</strong> tejidos) suma 282, mientras que el número <strong>de</strong> genes distintos es <strong>de</strong> 270. El<br />
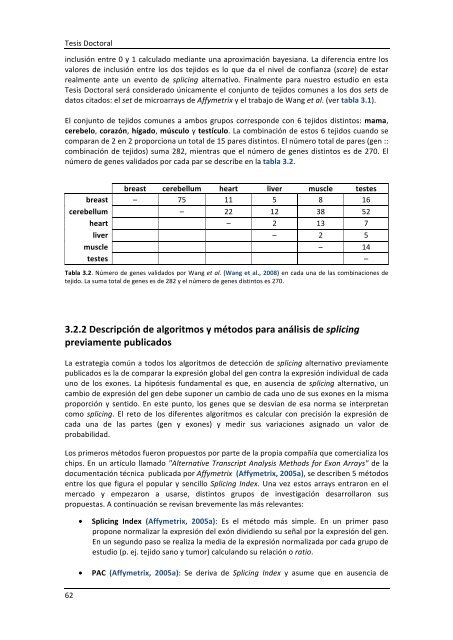
número <strong>de</strong> genes validados por cada par se <strong>de</strong>scribe en la tabla 3.2.<br />
breast cerebellum heart liver muscle testes<br />
breast – 75 11 5 8 16<br />
cerebellum – 22 12 38 52<br />
heart – 2 13 7<br />
liver – 2 5<br />
muscle – 14<br />
testes –<br />
Tabla 3.2. Número <strong>de</strong> genes validados por Wang et al. (Wang et al., 2008) en cada una <strong>de</strong> las combinaciones <strong>de</strong><br />
tejido. La suma total <strong>de</strong> genes es <strong>de</strong> 282 y el número <strong>de</strong> genes distintos es 270.<br />
3.2.2 Descripción <strong>de</strong> algoritmos y métodos para análisis <strong>de</strong> splicing<br />
previamente publicados<br />
La estrategia común a todos los algoritmos <strong>de</strong> <strong>de</strong>tección <strong>de</strong> splicing alternativo previamente<br />
publicados es la <strong>de</strong> comparar la expresión global <strong>de</strong>l gen contra la expresión individual <strong>de</strong> cada<br />
uno <strong>de</strong> los exones. La hipótesis fundamental es que, en ausencia <strong>de</strong> splicing alternativo, un<br />
cambio <strong>de</strong> expresión <strong>de</strong>l gen <strong>de</strong>be suponer un cambio <strong>de</strong> cada uno <strong>de</strong> sus exones en la misma<br />
proporción y sentido. En este punto, los genes que se <strong>de</strong>svían <strong>de</strong> esa norma se interpretan<br />
como splicing. El reto <strong>de</strong> los diferentes algoritmos es calcular con precisión la expresión <strong>de</strong><br />
cada una <strong>de</strong> las partes (gen y exones) y medir sus variaciones asignado un valor <strong>de</strong><br />
probabilidad.<br />
Los primeros métodos fueron propuestos por parte <strong>de</strong> la propia compañía que comercializa los<br />
chips. En un artículo llamado "Alternative Transcript Analysis Methods for Exon Arrays" <strong>de</strong> la<br />
documentación técnica publicada por Affymetrix (Affymetrix, 2005a), se <strong>de</strong>scriben 5 métodos<br />
entre los que figura el popular y sencillo Splicing In<strong>de</strong>x. Una vez estos arrays entraron en el<br />
mercado y empezaron a usarse, distintos grupos <strong>de</strong> investigación <strong>de</strong>sarrollaron sus<br />
propuestas. A continuación se revisan brevemente las más relevantes:<br />
62<br />
• Splicing In<strong>de</strong>x (Affymetrix, 2005a): Es el método más simple. En un primer paso<br />
propone normalizar la expresión <strong>de</strong>l exón dividiendo su señal por la expresión <strong>de</strong>l gen.<br />
En un segundo paso se realiza la media <strong>de</strong> la expresión normalizada por cada grupo <strong>de</strong><br />
estudio (p. ej. tejido sano y tumor) calculando su relación o ratio.<br />
• PAC (Affymetrix, 2005a): Se <strong>de</strong>riva <strong>de</strong> Splicing In<strong>de</strong>x y asume que en ausencia <strong>de</strong>













