

Alberto Risueño Pérez - Gredos - Universidad de Salamanca
Alberto Risueño Pérez - Gredos - Universidad de Salamanca
Alberto Risueño Pérez - Gredos - Universidad de Salamanca

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
Tesis Doctoral<br />
El mapeo <strong>de</strong> los HKG y <strong>de</strong> los TSG sobre esta red reveló también diferencias entre ambos<br />
métodos. El número <strong>de</strong> HKG mapeado sobre la red fue <strong>de</strong> 157 correspondiente a los genes<br />
presentes en al menos 3 <strong>de</strong> los 4 métodos propuestos. MAS5+Spearman ubicó en la red 77 <strong>de</strong><br />
estos genes mientras que RMA+Pearson, que es <strong>de</strong>masiado astringente, tan solo ubicó un 1<br />
HKG. Los TSG ubicados en la red se correspon<strong>de</strong>n con los i<strong>de</strong>ntificados (<strong>de</strong> nivel 1) en el<br />
apartado 4.2.4. En este caso RMA+Pearson fue más exitoso en la <strong>de</strong>tección <strong>de</strong> TSG <strong>de</strong>tectando<br />
351 por ninguno <strong>de</strong> MAS5+Spearman, consi<strong>de</strong>rando un total <strong>de</strong> 756 (ver tabla 4.5).<br />
94<br />
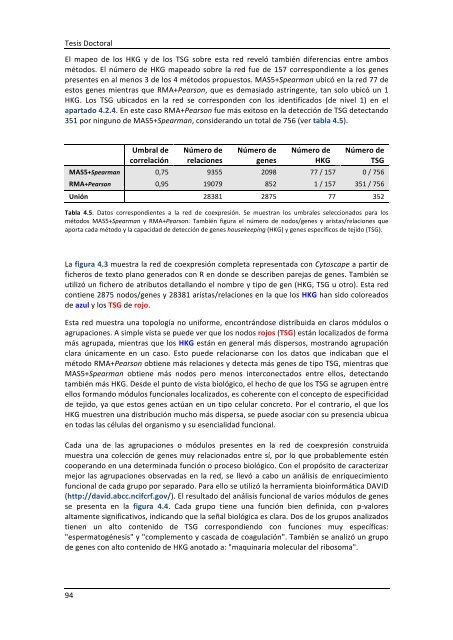
Umbral <strong>de</strong><br />
correlación<br />
Número <strong>de</strong><br />
relaciones<br />
Número <strong>de</strong><br />
genes<br />
Número <strong>de</strong><br />
HKG<br />
Número <strong>de</strong><br />
TSG<br />
MAS5+Spearman 0,75 9355 2098 77 / 157 0 / 756<br />
RMA+Pearson 0,95 19079 852 1 / 157 351 / 756<br />
Unión 28381 2875 77 352<br />
Tabla 4.5. Datos correspondientes a la red <strong>de</strong> coexpresión. Se muestran los umbrales seleccionados para los<br />
métodos MAS5+Spearman y RMA+Pearson. También figura el número <strong>de</strong> nodos/genes y aristas/relaciones que<br />
aporta cada método y la capacidad <strong>de</strong> <strong>de</strong>tección <strong>de</strong> genes housekeeping (HKG) y genes específicos <strong>de</strong> tejido (TSG).<br />
La figura 4.3 muestra la red <strong>de</strong> coexpresión completa representada con Cytoscape a partir <strong>de</strong><br />
ficheros <strong>de</strong> texto plano generados con R en don<strong>de</strong> se <strong>de</strong>scriben parejas <strong>de</strong> genes. También se<br />
utilizó un fichero <strong>de</strong> atributos <strong>de</strong>tallando el nombre y tipo <strong>de</strong> gen (HKG, TSG u otro). Esta red<br />
contiene 2875 nodos/genes y 28381 aristas/relaciones en la que los HKG han sido coloreados<br />
<strong>de</strong> azul y los TSG <strong>de</strong> rojo.<br />
Esta red muestra una topología no uniforme, encontrándose distribuida en claros módulos o<br />
agrupaciones. A simple vista se pue<strong>de</strong> ver que los nodos rojos (TSG) están localizados <strong>de</strong> forma<br />
más agrupada, mientras que los HKG están en general más dispersos, mostrando agrupación<br />
clara únicamente en un caso. Esto pue<strong>de</strong> relacionarse con los datos que indicaban que el<br />
método RMA+Pearson obtiene más relaciones y <strong>de</strong>tecta más genes <strong>de</strong> tipo TSG, mientras que<br />
MAS5+Spearman obtiene más nodos pero menos interconectados entre ellos, <strong>de</strong>tectando<br />
también más HKG. Des<strong>de</strong> el punto <strong>de</strong> vista biológico, el hecho <strong>de</strong> que los TSG se agrupen entre<br />
ellos formando módulos funcionales localizados, es coherente con el concepto <strong>de</strong> especificidad<br />
<strong>de</strong> tejido, ya que estos genes actúan en un tipo celular concreto. Por el contrario, el que los<br />
HKG muestren una distribución mucho más dispersa, se pue<strong>de</strong> asociar con su presencia ubicua<br />
en todas las células <strong>de</strong>l organismo y su esencialidad funcional.<br />
Cada una <strong>de</strong> las agrupaciones o módulos presentes en la red <strong>de</strong> coexpresión construida<br />
muestra una colección <strong>de</strong> genes muy relacionados entre sí, por lo que probablemente estén<br />
cooperando en una <strong>de</strong>terminada función o proceso biológico. Con el propósito <strong>de</strong> caracterizar<br />
mejor las agrupaciones observadas en la red, se llevó a cabo un análisis <strong>de</strong> enriquecimiento<br />
funcional <strong>de</strong> cada grupo por separado. Para ello se utilizó la herramienta bioinformática DAVID<br />
(http://david.abcc.ncifcrf.gov/). El resultado <strong>de</strong>l análisis funcional <strong>de</strong> varios módulos <strong>de</strong> genes<br />
se presenta en la figura 4.4. Cada grupo tiene una función bien <strong>de</strong>finida, con p-‐valores<br />
altamente significativos, indicando que la señal biológica es clara. Dos <strong>de</strong> los grupos analizados<br />
tienen un alto contenido <strong>de</strong> TSG correspondiendo con funciones muy específicas:<br />
"espermatogénesis" y "complemento y cascada <strong>de</strong> coagulación". También se analizó un grupo<br />
<strong>de</strong> genes con alto contenido <strong>de</strong> HKG anotado a: "maquinaria molecular <strong>de</strong>l ribosoma".













