

Alberto Risueño Pérez - Gredos - Universidad de Salamanca
Alberto Risueño Pérez - Gredos - Universidad de Salamanca
Alberto Risueño Pérez - Gredos - Universidad de Salamanca

Create successful ePaper yourself
Turn your PDF publications into a flip-book with our unique Google optimized e-Paper software.
Tesis Doctoral<br />
70<br />
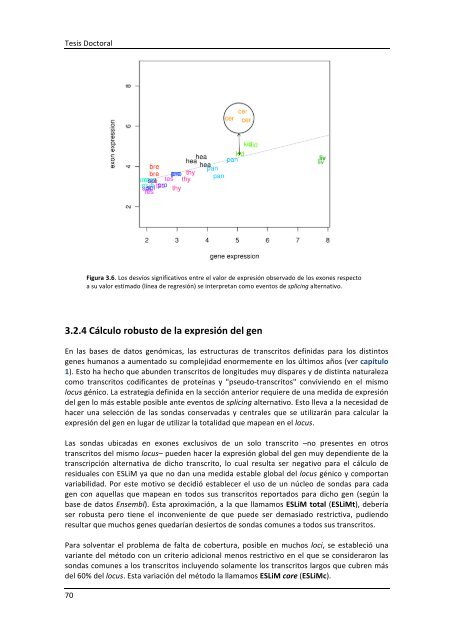
Figura 3.6. Los <strong>de</strong>svíos significativos entre el valor <strong>de</strong> expresión observado <strong>de</strong> los exones respecto<br />
a su valor estimado (línea <strong>de</strong> regresión) se interpretan como eventos <strong>de</strong> splicing alternativo.<br />
3.2.4 Cálculo robusto <strong>de</strong> la expresión <strong>de</strong>l gen<br />
En las bases <strong>de</strong> datos genómicas, las estructuras <strong>de</strong> transcritos <strong>de</strong>finidas para los distintos<br />
genes humanos a aumentado su complejidad enormemente en los últimos años (ver capítulo<br />
1). Esto ha hecho que abun<strong>de</strong>n transcritos <strong>de</strong> longitu<strong>de</strong>s muy dispares y <strong>de</strong> distinta naturaleza<br />
como transcritos codificantes <strong>de</strong> proteínas y "pseudo-‐transcritos" conviviendo en el mismo<br />
locus génico. La estrategia <strong>de</strong>finida en la sección anterior requiere <strong>de</strong> una medida <strong>de</strong> expresión<br />
<strong>de</strong>l gen lo más estable posible ante eventos <strong>de</strong> splicing alternativo. Esto lleva a la necesidad <strong>de</strong><br />
hacer una selección <strong>de</strong> las sondas conservadas y centrales que se utilizarán para calcular la<br />
expresión <strong>de</strong>l gen en lugar <strong>de</strong> utilizar la totalidad que mapean en el locus.<br />
Las sondas ubicadas en exones exclusivos <strong>de</strong> un solo transcrito –no presentes en otros<br />
transcritos <strong>de</strong>l mismo locus– pue<strong>de</strong>n hacer la expresión global <strong>de</strong>l gen muy <strong>de</strong>pendiente <strong>de</strong> la<br />
transcripción alternativa <strong>de</strong> dicho transcrito, lo cual resulta ser negativo para el cálculo <strong>de</strong><br />
residuales con ESLiM ya que no dan una medida estable global <strong>de</strong>l locus génico y comportan<br />
variabilidad. Por este motivo se <strong>de</strong>cidió establecer el uso <strong>de</strong> un núcleo <strong>de</strong> sondas para cada<br />
gen con aquellas que mapean en todos sus transcritos reportados para dicho gen (según la<br />
base <strong>de</strong> datos Ensembl). Esta aproximación, a la que llamamos ESLiM total (ESLiMt), <strong>de</strong>bería<br />
ser robusta pero tiene el inconveniente <strong>de</strong> que pue<strong>de</strong> ser <strong>de</strong>masiado restrictiva, pudiendo<br />
resultar que muchos genes quedarían <strong>de</strong>siertos <strong>de</strong> sondas comunes a todos sus transcritos.<br />
Para solventar el problema <strong>de</strong> falta <strong>de</strong> cobertura, posible en muchos loci, se estableció una<br />
variante <strong>de</strong>l método con un criterio adicional menos restrictivo en el que se consi<strong>de</strong>raron las<br />
sondas comunes a los transcritos incluyendo solamente los transcritos largos que cubren más<br />
<strong>de</strong>l 60% <strong>de</strong>l locus. Esta variación <strong>de</strong>l método la llamamos ESLiM core (ESLiMc).













