

Zalfa NOUR Modélisation de l'adsorption des molécules à fort ...
Zalfa NOUR Modélisation de l'adsorption des molécules à fort ...
Zalfa NOUR Modélisation de l'adsorption des molécules à fort ...

You also want an ePaper? Increase the reach of your titles
YUMPU automatically turns print PDFs into web optimized ePapers that Google loves.
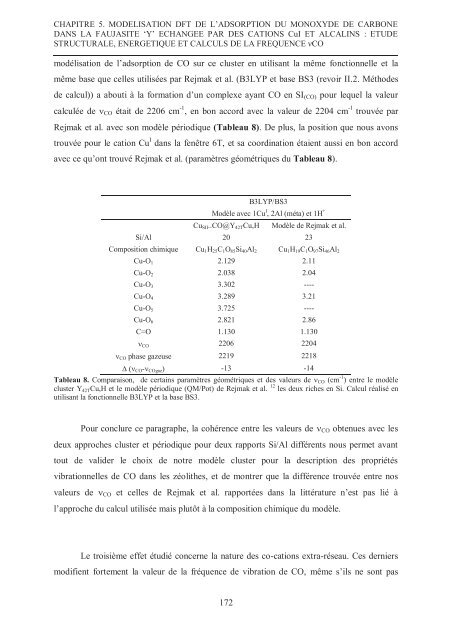
CHAPITRE 5. MODELISATION DFT DE L’ADSORPTION DU MONOXYDE DE CARBONEDANS LA FAUJASITE ‘Y’ ECHANGEE PAR DES CATIONS CuI ET ALCALINS : ETUDESTRUCTURALE, ENERGETIQUE ET CALCULS DE LA FREQUENCE νCOmodélisation <strong>de</strong> l’adsorption <strong>de</strong> CO sur ce cluster en utilisant la même fonctionnelle et lamême base que celles utilisées par Rejmak et al. (B3LYP et base BS3 (revoir II.2. Métho<strong>de</strong>s<strong>de</strong> calcul)) a abouti à la formation d’un complexe ayant CO en SI (CO) pour lequel la valeurcalculée <strong>de</strong> CO était <strong>de</strong> 2206 cm -1 , en bon accord avec la valeur <strong>de</strong> 2204 cm -1 trouvée parRejmak et al. avec son modèle périodique (Tableau 8). De plus, la position que nous avonstrouvée pour le cation Cu I dans la fenêtre 6T, et sa coordination étaient aussi en bon accordavec ce qu’ont trouvé Rejmak et al. (paramètres géométriques du Tableau 8).B3LYP/BS3Modèle avec 1Cu I , 2Al (méta) et 1H +Cu SII ..CO@Y 42T Cu,HModèle <strong>de</strong> Rejmak et al.Si/Al 20 23Composition chimique Cu 1 H 25 C 1 O 85 Si 40 Al 2 Cu 1 H 19 C 1 O 97 Si 46 Al 2Cu-O 1 2.129 2.11Cu-O 2 2.038 2.04Cu-O 3 3.302 ----Cu-O 4 3.289 3.21Cu-O 5 3.725 ----Cu-O 6 2.821 2.86C=O 1.130 1.130 CO 2206 2204 CO phase gazeuse 2219 2218∆ ( CO - COgaz ) -13 -14Tableau 8. Comparaison, <strong>de</strong> certains paramètres géométriques et <strong>de</strong>s valeurs <strong>de</strong> CO (cm -1 ) entre le modèlecluster Y 42T Cu,H et le modèle périodique (QM/Pot) <strong>de</strong> Rejmak et al. 12 les <strong>de</strong>ux riches en Si. Calcul réalisé enutilisant la fonctionnelle B3LYP et la base BS3.Pour conclure ce paragraphe, la cohérence entre les valeurs <strong>de</strong> CO obtenues avec les<strong>de</strong>ux approches cluster et périodique pour <strong>de</strong>ux rapports Si/Al différents nous permet avanttout <strong>de</strong> vali<strong>de</strong>r le choix <strong>de</strong> notre modèle cluster pour la <strong>de</strong>scription <strong>de</strong>s propriétésvibrationnelles <strong>de</strong> CO dans les zéolithes, et <strong>de</strong> montrer que la différence trouvée entre nosvaleurs <strong>de</strong> CO et celles <strong>de</strong> Rejmak et al. rapportées dans la littérature n’est pas lié àl’approche du calcul utilisée mais plutôt à la composition chimique du modèle.Le troisième effet étudié concerne la nature <strong>de</strong>s co-cations extra-réseau. Ces <strong>de</strong>rniersmodifient <strong>fort</strong>ement la valeur <strong>de</strong> la fréquence <strong>de</strong> vibration <strong>de</strong> CO, même s’ils ne sont pas172








