- Page 6:
Je présente aussi ma reconnaissanc
- Page 10 and 11:
Cas de la NaY :....................
- Page 12 and 13:
INTRODUCTION GENERALELa nanotechnol
- Page 14 and 15:
INTRODUCTION GENERALEdifférents mo
- Page 16:
INTRODUCTION GENERALEcours de cette
- Page 19 and 20:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 21 and 22:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 23 and 24:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 25 and 26:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 27 and 28:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 29 and 30:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 31 and 32:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 33 and 34:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 35 and 36:
CHAPITRE 1. LES ZEOLITHES : PRESENT
- Page 37 and 38:
25. Maurin, G.; Plant, D. F.; Henn,
- Page 39 and 40:
and Catalysis 1994, 84, (ZEOLITES A
- Page 41 and 42:
III.2.4 Implémentation d’une sim
- Page 44 and 45:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 46 and 47:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 48 and 49:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 50 and 51:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 52 and 53:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 54 and 55:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 56 and 57:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 58 and 59:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 60 and 61:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 62 and 63:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 64 and 65:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 66 and 67:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 68 and 69:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 70 and 71:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 72 and 73:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 74 and 75:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 76 and 77:
CHAPITRE 2. NOTIONS FONDAMENTALES S
- Page 78 and 79:
Références bibliographiques1. Pau
- Page 80 and 81:
39. Jeffroy, M. Simulation Molécul
- Page 82 and 83:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 84 and 85:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 86 and 87:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 88 and 89:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 90 and 91:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 92 and 93:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 94 and 95:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 96 and 97:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 98 and 99:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 100 and 101:
CHAPITRE 3. NOTIONS FONDAMENTALES S
- Page 102 and 103:
Références Bibliographiques1. Cra
- Page 104 and 105:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 106 and 107:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 108 and 109:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 110 and 111:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 112 and 113:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 114 and 115:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 116 and 117:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 118 and 119:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 120 and 121:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 122 and 123:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 124 and 125:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 126 and 127:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 128 and 129:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 130 and 131:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 132 and 133:
CHAPITRE 4. ETUDE BIBLIOGRAPHIQUE D
- Page 134 and 135:
18. Zecchina, A.; Bordiga, S.; Turn
- Page 136 and 137:
50. Rejmak, P.; Sierka, M.; Sauer,
- Page 138 and 139:
VI. ANNEXES .......................
- Page 140 and 141:
CHAPITRE 5. MODELISATION DFT DE L
- Page 142 and 143:
CHAPITRE 5. MODELISATION DFT DE L
- Page 144 and 145:
CHAPITRE 5. MODELISATION DFT DE L
- Page 146 and 147:
CHAPITRE 5. MODELISATION DFT DE L
- Page 148 and 149:
CHAPITRE 5. MODELISATION DFT DE L
- Page 150 and 151:
CHAPITRE 5. MODELISATION DFT DE L
- Page 153 and 154:
CHAPITRE 5. MODELISATION DFT DE L
- Page 155 and 156:
Tableau 5. Paramètres géométriqu
- Page 157 and 158:
CHAPITRE 5. MODELISATION DFT DE L
- Page 159 and 160:
CHAPITRE 5. MODELISATION DFT DE L
- Page 161 and 162:
Tableau 6. Paramètres géométriqu
- Page 163 and 164:
CHAPITRE 5. MODELISATION DFT DE L
- Page 165 and 166: CHAPITRE 5. MODELISATION DFT DE L
- Page 167 and 168: CHAPITRE 5. MODELISATION DFT DE L
- Page 169 and 170: CHAPITRE 5. MODELISATION DFT DE L
- Page 171 and 172: CHAPITRE 5. MODELISATION DFT DE L
- Page 173 and 174: CHAPITRE 5. MODELISATION DFT DE L
- Page 175 and 176: CHAPITRE 5. MODELISATION DFT DE L
- Page 177 and 178: CHAPITRE 5. MODELISATION DFT DE L
- Page 179 and 180: CHAPITRE 5. MODELISATION DFT DE L
- Page 181 and 182: CHAPITRE 5. MODELISATION DFT DE L
- Page 183 and 184: CHAPITRE 5. MODELISATION DFT DE L
- Page 185 and 186: CHAPITRE 5. MODELISATION DFT DE L
- Page 187 and 188: CHAPITRE 5. MODELISATION DFT DE L
- Page 189 and 190: CHAPITRE 5. MODELISATION DFT DE L
- Page 191 and 192: Avec ces valeurs nous avons obtenu
- Page 193 and 194: Références Bibliographiques1. Ber
- Page 195 and 196: 32. Berthomieu, D.; Krishnamurty, S
- Page 197 and 198: 66. Kozyra, P.; Salla, I.; Montanar
- Page 199 and 200: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 201 and 202: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 203 and 204: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 205 and 206: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 207 and 208: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 209 and 210: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 211 and 212: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 213 and 214: Tableau 5. Comparaison des paramèt
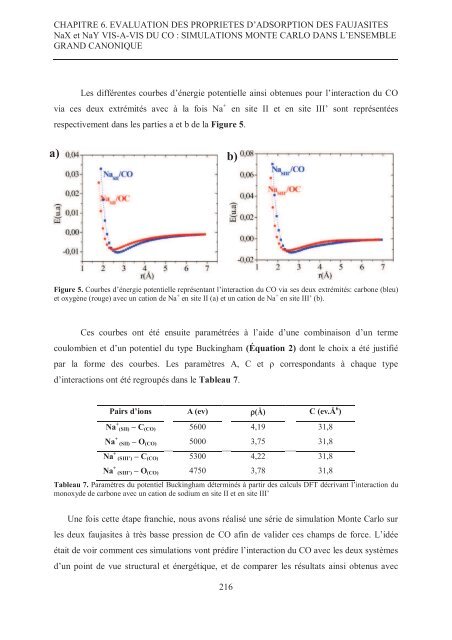
- Page 215: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 219 and 220: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 221 and 222: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 223 and 224: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 225 and 226: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 227 and 228: CHAPITRE 6. EVALUATION DES PROPRIET
- Page 229 and 230: Références bibliographiques1. Par
- Page 231 and 232: 33. Zecchina, A.; Otero Arean, C.;
- Page 233 and 234: 61. Soltanov, R. I.; Paukstis, E.;
- Page 235 and 236: CONCLUSION GENERALEDifférents aspe