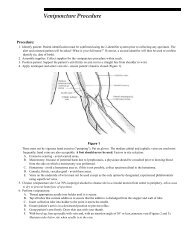
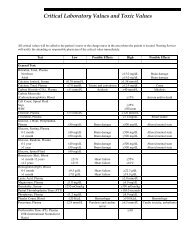
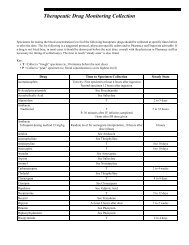
- Page 1 and 2:
Rochester 2013 Interpretive Handboo
- Page 3 and 4:
Policies - Mayo Medical Laboratorie
- Page 5 and 6:
Policies - Mayo Medical Laboratorie
- Page 7 and 8:
Policies - Mayo Medical Laboratorie
- Page 9 and 10:
Policies - Mayo Medical Laboratorie
- Page 11 and 12:
Policies - Mayo Medical Laboratorie
- Page 13 and 14:
TTIG 82506 DHVD 8822 Tetanus Toxoid
- Page 15 and 16:
DCRN 8847 Identification of patient
- Page 17 and 18:
DOC 8547 function primarily, 18-hyd
- Page 19 and 20:
FDSOX 91690 THCM 84284 11-Desoxycor
- Page 21 and 22:
FBP1 86208 Normal: or =1.5 ng/mL;
- Page 23 and 24:
17OHP 81151 Pediatric Reference Ran
- Page 25 and 26:
OHPG 9231 2000;52(5):601-607 4. Kao
- Page 27 and 28:
FP73 88541 Interpretation: The pres
- Page 29 and 30:
OH21 81970 21-deoxycortisol may be
- Page 31 and 32:
CYPKP 89082 transcriptionally activ
- Page 33 and 34:
25HDN 83670 25-Hydroxyvitamin D2 an
- Page 35 and 36:
F5HAR 57333 HIAA 9248 can also be a
- Page 37 and 38:
6MAMU 89605 converts heroin into 6-
- Page 39 and 40:
ACAC 82757 ACANT 80401 increase the
- Page 41 and 42:
ACM 8698 FACTO 90247 1 0.35-0.69 Eq
- Page 43 and 44:
ACHS 8522 Clinical Information: Neu
- Page 45 and 46:
ACT 8221 APT 9058 and isolation of
- Page 47 and 48:
AHPS 9022 ancestry. Homozygosity fo
- Page 49 and 50:
FAML secretions, but it is not comm
- Page 51 and 52:
ACRN 82413 Acylcarnitines, Quantita
- Page 53 and 54:
or =8 years:
- Page 55 and 56:
ADM13 61212 studies (enzyme assay,
- Page 57 and 58:
FADA 91554 81444 (50-70 x normal) A
- Page 59 and 60:
FADE 91670 LADV 89074 LCADP 89887 A
- Page 61 and 62:
FADMK 91925 RACTH 82140 ADmark Phos
- Page 63 and 64:
AGXMS 89915 7 year 2-88 8 year 5-71
- Page 65 and 66:
the third decade of life, but can o
- Page 67 and 68:
FALUF 57286 ALB 8436 involved in th
- Page 69 and 70:
FALCO 90084 ALS 8363 ALDNA 15150 th
- Page 71 and 72:
ALDU 8556 regulator of the synthesi
- Page 73 and 74:
ALKI 89503 11 years: 185-507 U/L 12
- Page 75 and 76:
17-23 years: 52-144 U/L 24-45 years
- Page 77 and 78:
FALMD 92001 ALM 82882 Almond (Amygd
- Page 79 and 80:
FASU 91221 FA1AG 90464 19-70+ years
- Page 81 and 82:
A1APP 26953 genotype is at greater
- Page 83 and 84:
A1M24 81036 RA1M 84448 605-608 2. M
- Page 85 and 86:
A2M 9270 AAMY 82866 Alpha-2-Macrogl
- Page 87 and 88:
AFP 8162 hepatocellular carcinoma,
- Page 89 and 90:
AFPSF 8876 Screen [Second Trimester
- Page 91 and 92:
FUCW 8814 molecules in the tissues
- Page 93 and 94:
AGA 8785 disease. In female patient
- Page 95 and 96:
AGPB 9499 IDSWB 60618 disease. In T
- Page 97 and 98:
IDST 8780 categorized into 3 syndro
- Page 99 and 100:
MANT 8773 MANN 8772 5 50.0-99.9 Str
- Page 101 and 102:
ANAS 8782 APGH 9003 Alpha-N-Acetylg
- Page 103 and 104:
ALPA 500155 Tanner II-IV*: < or =1.
- Page 105 and 106:
ALUR 86377 -Aluminum-laden albumin
- Page 107 and 108:
86375 exposure. This was done by sw
- Page 109 and 110:
FAMAN 91132 AMKPK 82112 possible ev
- Page 111 and 112:
AAMSD 60200 susceptible strains of
- Page 113 and 114:
Serine (Ser) 69-271 71-208 63-187 H
- Page 115 and 116:
Glutamine Gln 139-2985 263 -2979 15
- Page 117 and 118:
AAUCD 60202 Glutamic Acid (Glu) 1-M
- Page 119 and 120:
FALAU 57350 ALADW to secondary inhi
- Page 121 and 122:
AMIO 9247 dehydratase (ALAD) activi
- Page 123 and 124:
AMOBS 8325 FAMOX 80450 FMAXN 57245
- Page 125 and 126:
AMPCC 61514 receptors that mediate
- Page 127 and 128:
FAMPH 90113 AMPHU 8257 MJ, Parks PM
- Page 129 and 130:
AMBF 8371 immune response to allerg
- Page 131 and 132:
PAMYB 5079 PAMY 80376 Clinical Refe
- Page 133 and 134:
AMSU 8356 AMS 8352 Amylase, Timed C
- Page 135 and 136:
AMYL 83667 AMYKM 83705 provided. Re
- Page 137 and 138:
ANAP 81157 TTR-associated familial
- Page 139 and 140:
F3AAG 57109 ANST 9709 Class IgE kU/
- Page 141 and 142:
FPOC 81081 correlate only modestly
- Page 143 and 144:
FANGI 90429 FANG 90428 Interpretati
- Page 145 and 146:
FANSD 91955 FAEAB 91854 immunoglobu
- Page 147 and 148:
FANBF 57173 FCLNE 91321 FPHET 91322
- Page 149 and 150:
ABTIH 9000 ENAE 89035 Antibody Tite
- Page 151 and 152:
MMLYP 81602 MMLRG 81601 Test Perfor
- Page 153 and 154:
MMLSA 56031 MACCL 89218 antimicrobi
- Page 155 and 156:
FAMS 91540 Clinical Information: No
- Page 157 and 158:
VASC 83012 evaluation for infertili
- Page 159 and 160:
ANA2 9026 ATTF 9030 Antinuclear Ant
- Page 161 and 162:
ATTI 9031 Antithrombin Antigen, Pla
- Page 163 and 164:
APO1K 60724 APO2S 60725 Shiller SM,
- Page 165 and 166:
APLAB 80318 APLA1 80309 mutation in
- Page 167 and 168:
APOE 80905 expressing more severe d
- Page 169 and 170:
APR 82835 AWNS 87814 6 > or =100 St
- Page 171 and 172:
ARBOP 83267 Reference values apply
- Page 173 and 174:
distinguishable from comparison gro
- Page 175 and 176:
FARI 57112 FARIX 57123 FCGHP 89858
- Page 177 and 178:
ARSAS 61259 ARSAK number variation
- Page 179 and 180:
ASFRU 84679 forms. Concentrations >
- Page 181 and 182:
ASHA 8651 and their partially detox
- Page 183 and 184:
ASCRU 89890 are nontoxic, inorganic
- Page 185 and 186:
ARSAW 8779 Useful For: Detection of
- Page 187 and 188:
ARSB 8151 ASCRI 82764 Roth. McGraw-
- Page 189 and 190:
AJPWO 88887 Clinical References: 1.
- Page 191 and 192:
AST 8360 clinical manifestations. I
- Page 193 and 194:
FASP 91607 ASPBA 61009 antigen in f
- Page 195 and 196:
SASP 9678 ASPG 86324 Aspergillus fu
- Page 197 and 198:
FAPP 91428 Reference Values: or =1
- Page 199 and 200:
APIN 82803 responsible for elicitin
- Page 201 and 202:
AGIDE 89886 < or =0.02 nmol/L NEURO
- Page 203 and 204:
ALDE 84248 anti-neuronal nuclear au
- Page 205 and 206:
FAVI 91509 AVOC 82812 approximately
- Page 207 and 208:
CD40 89009 conventional cytogenetic
- Page 209 and 210:
80027 IABCS 88800 patients with B-c
- Page 211 and 212:
B-cell-depleting immunotherapy Iden
- Page 213 and 214:
functional classification system fo
- Page 215 and 216:
FBAB 91608 BABG 81128 5785 Corporat
- Page 217 and 218:
GEN 8108 SPUT 8095 Bruyn G, Pieniaz
- Page 219 and 220:
CFRC 89653 EPRP 60235 result in int
- Page 221 and 222:
BAHG 82711 BCYP 82722 electrophores
- Page 223 and 224:
BANA 82746 sensitization to particu
- Page 225 and 226:
BRLY 82687 caused by the release of
- Page 227 and 228:
FBART 91439 BART 81575 amplificatio
- Page 229 and 230:
BARTB 89983 BASL 82489 B. bacillifo
- Page 231 and 232:
BADX 89006 Interpretation: Detectio
- Page 233 and 234:
MBCR 80578 helpful for both prognos
- Page 235 and 236:
FBBCK 91664 FBEAN 91646 ASPE/Lumine
- Page 237 and 238:
BECH 82669 of IC2 (LIT1) is hypothe
- Page 239 and 240:
BEEF 82697 BEETS 82618 Beef, IgE Cl
- Page 241 and 242:
FPHEN 91136 FBEN 90294 BENZU 80370
- Page 243 and 244:
BBEET 82838 BERG 82892 Berlin Beetl
- Page 245 and 246:
AB2GP 86180 Negative (reported as p
- Page 247 and 248:
GB2GP 86182 or =15.0 U/mL (positiv
- Page 249 and 250:
BETA2 80351 patients with APS.(4) A
- Page 251 and 252:
B2MU 300243 B2M 9234 Livanainen M,
- Page 253 and 254:
BGAW 60987 Postmenopausal: 104-1,00
- Page 255 and 256:
BGAT 8008 beta-galactosidase defici
- Page 257 and 258:
BGLT 8787 BGL 8788 B disease. Clini
- Page 259 and 260:
BHCG 61718 Sly syndrome is 1 of the
- Page 261 and 262:
BLACT 8118 BLAC Clinical Informatio
- Page 263 and 264:
and the determination of bicarbonat
- Page 265 and 266:
84357 19701 Interpretation: Total b
- Page 267 and 268:
BFBL 34621 the color of bilirubin a
- Page 269 and 270:
BILIT 81785 Bilirubin, Total, Serum
- Page 271 and 272:
BIOTS 88205 levels of biotinidase,
- Page 273 and 274:
FBISU 91142 LCBK 88910 Test Perform
- Page 275 and 276:
QBKU 87859 Useful For: As a prospec
- Page 277 and 278:
SBLAS 86691 antibodies interact wit
- Page 279 and 280:
80326 BDIAL 83094 night sweats. It
- Page 281 and 282:
UEBF 81834 BWOR 82840 urinary tract
- Page 283 and 284:
BLUE 82359 Clinical Information: Cl
- Page 285 and 286:
BHINT 9027 resorption is balanced b
- Page 287 and 288:
BPRP 80910 FBPTS 57290 An interpret
- Page 289 and 290:
BOAC 9723 BOT 82715 Cypress, CA 906
- Page 291 and 292:
89045 proteins) followed by respira
- Page 293 and 294:
BRAZ 82899 MLH1 promoter hypermethy
- Page 295 and 296:
BMNA 81976 FYSTB 91990 BROC 82817 C
- Page 297 and 298:
BRM 8608 BRUGM 89476 0 Negative 1 0
- Page 299 and 300:
BRUTA 8112 brucellosis may include
- Page 301 and 302:
manifests in male children younger
- Page 303 and 304:
clinical phenotype can vary conside
- Page 305 and 306:
BTKK 89306 Useful For: Confirming a
- Page 307 and 308:
BUCW 82727 XLA patients are diagnos
- Page 309 and 310:
BFTH 82779 identify allergens which
- Page 311 and 312:
BUPIS 89548 BUPM 500038 membrane pr
- Page 313 and 314:
FBUS 91115 BUAUC 83188 increase in
- Page 315 and 316:
FCPEP 91270 anti-insulin autoantibo
- Page 317 and 318:
C1ES 8198 Useful For: Assessment of
- Page 319 and 320:
C1QFX 83374 Reference Values: C1Q B
- Page 321 and 322:
C2 81835 receptors. The absence of
- Page 323 and 324:
FC3D 91725 C4U 88829 C4FX 83391 C3d
- Page 325 and 326:
C5FX 83392 C5DCU 88831 1987;76:939-
- Page 327 and 328:
C7FX 81064 complement system is com
- Page 329 and 330:
C9FX 81066 CABB 86327 4. Gaither TA
- Page 331 and 332:
CDOMB 89539 CDOM 80595 Reference Va
- Page 333 and 334:
CDRU 60156 CAFF 8754 years) is the
- Page 335 and 336:
3 10.7-15.4 17-72 4 11.8-16.2 15-11
- Page 337 and 338:
FCAH1 91275 1 - 7m: Levels decrease
- Page 339 and 340:
Males: Levels increase after the fi
- Page 341 and 342:
Units: ug/dL Age Range Premature (2
- Page 343 and 344:
4 10.7-15.6 47-208 5 11.8-18.6 50-2
- Page 345 and 346:
anging from 40 - 200 between 30 and
- Page 347 and 348:
Clinical Information: In the normal
- Page 349 and 350:
CSRMS 83703 hypoparathyroidism, a s
- Page 351 and 352:
CAU 8594 CAF 8379 An interpretive r
- Page 353 and 354:
CAUR 60157 CAS parathyroid hormone-
- Page 355 and 356:
CACRU 89604 important role in blood
- Page 357 and 358:
CAVPC 83900 excretion of calcium is
- Page 359 and 360:
FCALP 91597 FCAMP 91224 CFTH 82778
- Page 361 and 362:
CANW 81780 CA25 9289 1 0.35-0.69 Eq
- Page 363 and 364:
FCANA 57158 testing often depend up
- Page 365 and 366:
FFTH 90479 FCAPR 90062 CWAY 82493 C
- Page 367 and 368:
CAR 8654 C1011 80682 total and free
- Page 369 and 370:
199PC 89508 199PT 61530 Clinical Re
- Page 371 and 372:
CA19 9288 CDG 89891 Carbohydrate An
- Page 373 and 374:
CHOU 9255 COHBB 8649 isoforms (ie,
- Page 375 and 376:
CEAPT 61528 PFCEA 83742 Carcinoembr
- Page 377 and 378:
CEASF 8918 CARD 82491 Carcinoembryo
- Page 379 and 380:
CPTKM 61121 CARN 8802 screening can
- Page 381 and 382:
cycle, or secondary disturbances in
- Page 383 and 384:
CACTK 61195 CAROB 82368 analysis. U
- Page 385 and 386:
FCASE 91647 caused by the release o
- Page 387 and 388:
FCASO 91995 CBN 82770 sensitivity t
- Page 389 and 390:
COMTO 60336 1 0.35-0.69 Equivocal 2
- Page 391 and 392:
CATU 9276 An interpretive report wi
- Page 393 and 394:
neuropathies are characterized by e
- Page 395 and 396:
CBC 9109 Useful For: Testing for Ig
- Page 397 and 398:
15 days-1 month: 31.0-55.0% 2-5 mon
- Page 399 and 400:
CD20B 89584 CD20 on B Cells Clinica
- Page 401 and 402:
3-5 months: 51-77%* 6-11 months: 49
- Page 403 and 404:
55 years: 49-87% % Helper cells (CD
- Page 405 and 406:
GLICP 89369 essential to serially m
- Page 407 and 408:
T- AND B-CELL QUANTITATION BY FLOW
- Page 409 and 410:
GLIC 89317 H/S ratio: > or =1.0 *Sh
- Page 411 and 412:
a more recent thymic ontogeny where
- Page 413 and 414:
CDKKM 60229 Med Genet 1999;36:518-5
- Page 415 and 416:
CELY 82766 CDCOM 89201 Company, 200
- Page 417 and 418:
CDGF 89200 serology does not exclud
- Page 419 and 420:
CELI 88906 range. For these individ
- Page 421 and 422:
5368 CMA 9278 autoantibodies can be
- Page 423 and 424:
CTSA 81979 sensitization to particu
- Page 425 and 426:
CERE 8364 patients with multiple sc
- Page 427 and 428:
8032 80199 2002 April ;287(16):2114
- Page 429 and 430:
CFTRK 88880 deferens or pancreatiti
- Page 431 and 432:
CCHZ 82752 MCHZ 82751 Test Performe
- Page 433 and 434:
CTRE 82607 likelihood of allergic d
- Page 435 and 436:
CHXP 82494 Clinical Information: Im
- Page 437 and 438:
CHCK 82713 CSPR 82351 5 50.0-99.9 S
- Page 439 and 440:
CHILI 82499 and to define the aller
- Page 441 and 442:
CHRGB 83186 CHPA 80411 FCPP 57339 S
- Page 443 and 444:
CGRNA 61553 Clinical References: 1.
- Page 445 and 446:
FCTRC 91659 CDFAW 110502 are requir
- Page 447 and 448:
FCPC 90439 FCPD 91750 FCHH 90111 pr
- Page 449 and 450:
CLFT 60028 CLBF 8470 CLF 8467 as hi
- Page 451 and 452:
CLSF 8218 CLU 8531 FCCK 90162 > or
- Page 453 and 454:
HDCH 8429 CHOL 8320 Serous Body Flu
- Page 455 and 456:
BHSF 8877 FCHAB 91900 serum may inc
- Page 457 and 458:
CR 86153 concentrations in the abse
- Page 459 and 460:
(5-hydroxytryptamine: 5-HT) and pep
- Page 461 and 462:
POC 8887 CVS 80257 An interpretativ
- Page 463 and 464:
CMS 8696 HBL 8537 deletions in 13q1
- Page 465 and 466:
LN 8911 SCE 926 Chromosome Analysis
- Page 467 and 468:
SBK 89338 to summarize here. The re
- Page 469 and 470:
CHSBP 9023 process. A normal karyot
- Page 471 and 472:
FCLL 83089 Negative Interpretation
- Page 473 and 474:
CHYM 82609 the urine (chyluria) is
- Page 475 and 476:
CTCB 89089 CTCC 89162 This test was
- Page 477 and 478:
RCITR 84773 CITR 9329 Reference Val
- Page 479 and 480:
CLAM 82884 proteins) followed by re
- Page 481 and 482:
CLLM 60490 Reference Values: An int
- Page 483 and 484:
CLOS 80424 CDRP 83124 Sedation has
- Page 485 and 486:
FCMVQ 91734 recurrent suicidal beha
- Page 487 and 488:
F_9 9065 FACTV 9054 Adults: 75-145%
- Page 489 and 490:
F7IS 7809 F8A 9070 prolonged prothr
- Page 491 and 492:
FXCH 89042 F10IS 7811 Coagulation F
- Page 493 and 494:
F_12 9069 COU 80083 Basic Principle
- Page 495 and 496:
COS 80084 COBCU 60353 Cobalt, Serum
- Page 497 and 498:
COKEU 9286 fluid.(7) The first evid
- Page 499 and 500:
CCOC 81542 Interpretation: Compleme
- Page 501 and 502:
COCR 82693 clinical manifestations.
- Page 503 and 504:
COD 82889 Q10 87853 5 50.0-99.9 Str
- Page 505 and 506:
CAGG 8992 FFTYC 91496 CTF 80440 Col
- Page 507 and 508:
CVID 87993 testing often depend upo
- Page 509 and 510:
C4 8171 AH50 88676 Clinical Informa
- Page 511 and 512:
FCCEV 57461 FFDMC 91581 FMDMC 91578
- Page 513 and 514:
CAH21 87815 congenital adrenal hype
- Page 515 and 516:
CTDC 83631 Reference Values: An int
- Page 517 and 518:
CURU Interpretation: The constellat
- Page 519 and 520:
CUCRU 60427 CORI can cause hypocupr
- Page 521 and 522:
CRNP 82718 CORN 82705 Corn Pollen,
- Page 523 and 524:
sex steroids. Synthesis proceeds fr
- Page 525 and 526:
FCBG 90148 FCORT 91644 CRANU 82920
- Page 527 and 528:
CORTU 8546 Cortisol, Serum (ug/dL)
- Page 529 and 530:
CORT 8545 measurements, midnight bl
- Page 531 and 532:
COCRU 88903 serum and urine cortiso
- Page 533 and 534:
hydrocortisone) increases and is fi
- Page 535 and 536:
CSED 82804 Class IgE kU/L Interpret
- Page 537 and 538:
FACX 91340 immune response to aller
- Page 539 and 540:
89366 CPOXS 61263 CPM, 12q15, for W
- Page 541 and 542:
CRANB 86307 bronchospasm) in infant
- Page 543 and 544:
CRDPU 88697 6 > or =100 Strongly po
- Page 545 and 546:
CKMB 82429 CK 8336 Creatine Kinase
- Page 547 and 548:
CREAZ 8472 13-36 months: 0.1-0.4 mg
- Page 549 and 550:
CRBF 8037 RCTUR 83802 guidelines fo
- Page 551 and 552:
CRGSP 83659 CRY_S 80988 Clinical Re
- Page 553 and 554:
CCRYP 86166 CRYPF 60320 in serum sp
- Page 555 and 556:
CRYPU 60319 Clinical Information: C
- Page 557 and 558:
CUKE 82861 Reference Values: Anti-T
- Page 559 and 560:
WHTC 82915 proteins) followed by re
- Page 561 and 562:
VRID2 5190 FCMSD 92004 CURR 82498 C
- Page 563 and 564:
60506 CIFS 8052 Cutaneous Anaplasti
- Page 565 and 566:
CYAN 8691 GRP 8771 pattern at the B
- Page 567 and 568:
CYCL 81506 CYCSP 8931 Reference Ran
- Page 569 and 570:
insufficiency. The incidence of CF
- Page 571 and 572:
CYSR 81067 distinguished by the pat
- Page 573 and 574:
drugs and environmental factors. Us
- Page 575 and 576:
2C19S 60439 activity include: -Broc
- Page 577 and 578:
including the intestines and liver.
- Page 579 and 580:
2C9SO 60337 Interpretation: An inte
- Page 581 and 582:
2613delAGA Decreased activity *10 1
- Page 583 and 584:
2D6TO 60340 an interpretation indic
- Page 585 and 586:
antidepressants, antiemetics, antih
- Page 587 and 588:
3A4O 61242 individual is homozygous
- Page 589 and 590:
CMG 80750 known. A ratio of > or =2
- Page 591 and 592:
e seen after acute illness, immunos
- Page 593 and 594:
and have cytotoxic function in resp
- Page 595 and 596:
ANCA 9441 Clinical Information: Cyt
- Page 597 and 598:
DLAC 8878 DLAU 8873 fibrinogen equi
- Page 599 and 600:
DAND 82694 FDANT 90363 5 50.0-99.9
- Page 601 and 602:
DATRE 82481 9803 DEEP 82144 Date, T
- Page 603 and 604:
observed at birth. Levels then decl
- Page 605 and 606:
diagnostic accuracy than DHEA-S mea
- Page 607 and 608:
FDOC 90134 5468 children. Character
- Page 609 and 610:
DESIP 81854 DSG13 83680 1 0.35-0.69
- Page 611 and 612:
FDXM 91956 FDEXA 91777 diagnosis, t
- Page 613 and 614:
DIA 8629 FDIGS 91454 Clinical Refer
- Page 615 and 616:
DIG 8674 FDPD 57141 American Associ
- Page 617 and 618:
DILL 82602 serum levels. Patients w
- Page 619 and 620:
DIP 83262 DTAB 83269 FDIPY 90371 Co
- Page 621 and 622:
FDISP 91595 DSP 8220 Sucrase: Range
- Page 623 and 624:
FDKYL 91960 DOGD 60108 Increases of
- Page 625 and 626:
DRD3O 60342 should be monitored clo
- Page 627 and 628:
DRD4O 60344 Reference Values: An in
- Page 629 and 630:
FDOXY 90061 CDAU1 80917 doxepin and
- Page 631 and 632:
CDAU3 80919 spectrometry (GC-MS) th
- Page 633 and 634:
IDOAU 8248 (GC-FID) the following d
- Page 635 and 636:
DABAR 505335 Urine Clinical Informa
- Page 637 and 638:
DMETH 505343 DPCP 505339 EMIT cutof
- Page 639 and 640:
DTHC 505331 and kidney. Toxic manif
- Page 641 and 642:
DSS 8421 CDAS 500752 Drugs of toxic
- Page 643 and 644:
FBDAS 91776 FMP10 57505 FBD 57117 D
- Page 645 and 646:
DASM4 60553 spectrometry (LC-MS/MS)
- Page 647 and 648:
DULOX 89305 immunoglobulin E (IgE)-
- Page 649 and 650:
ESYC 82721 infected individuals, it
- Page 651 and 652:
FECHC 91342 ECHO 80293 concentratio
- Page 653 and 654:
FEGFR 91903 FEGGW 91976 EGG 82872 W
- Page 655 and 656:
EGGP 82477 concentration of IgE ant
- Page 657 and 658:
FECHA 91710 EHRL 84319
- Page 659 and 660:
ELDR 82392 ELPN 87972 No pediatric
- Page 661 and 662:
EFP 81488 SODIUM 0-15 years: not es
- Page 663 and 664:
PEL 80085 chain fragments as well a
- Page 665 and 666:
ELM 82672 PROTEIN, TOTAL > or =18 y
- Page 667 and 668:
Reference Range: IgG
- Page 669 and 670:
5362 FEMA 91836 EMA 9360 anatomic p
- Page 671 and 672:
SAM 9049 FEHA 91932 Interpretation:
- Page 673 and 674:
FENTQ 91312 LENT 80066 INTERPRETIVE
- Page 675 and 676:
FEPHD 90109 EPUR 82854 presence of
- Page 677 and 678:
SEBV 84421 Clinical Information: Cl
- Page 679 and 680:
80786 EBVA 8891 antigen. The presen
- Page 681 and 682:
LEBV 81239 EBVB 87439 with type 1 N
- Page 683 and 684:
REVP 84160 M, Emre S, et al: Prospe
- Page 685 and 686:
EPOR 61679 FESC 91458 FFES no eryth
- Page 687 and 688:
primarily in ovaries and testes by
- Page 689 and 690:
UE3 81711 Stage V 18 years 10-40 pg
- Page 691 and 692:
ESTF 84230 Immunohistochemistry, Ma
- Page 693 and 694:
preparations). The gonadotrophin-re
- Page 695 and 696:
E1 81418 Stage V 14.5 years 15-350
- Page 697 and 698:
are due to problems in androgen sig
- Page 699 and 700:
ETOHU 500323 ETX 8769 Interpretatio
- Page 701 and 702:
EOXD 82767 EUCL 82758 Ethylene Oxid
- Page 703 and 704:
EHOR 82662 0 Negative 1 0.35-0.69 E
- Page 705 and 706:
83363 fluorescence in situ hybridiz
- Page 707 and 708:
F9INH 83103 lysosomes of both perip
- Page 709 and 710:
F8INH 83102 FX13M 57302 FOGT Clinic
- Page 711 and 712:
FAPKM 83001 and to define the aller
- Page 713 and 714:
FD 85319 LDLRS 81013 features This
- Page 715 and 716:
LDLM 89073 Useful For: Genetic test
- Page 717 and 718:
FANCA 85318 FATF 8310 separately or
- Page 719 and 720:
FAPCP 82042 Clinic or elsewhere, an
- Page 721 and 722:
or =18 years: 30-450 nmol/mL Hexade
- Page 723 and 724:
Hexacosanoic Acid, C26:0 0.00-1.30
- Page 725 and 726:
1-17 years: 9-130 nmol/mL > or =18
- Page 727 and 728:
FAPM 81939 or =18 years: 7.3-16.8
- Page 729 and 730:
POX 81369 oxidation disorders. Ann
- Page 731 and 732:
FBN1 89308 range of variability, in
- Page 733 and 734:
FETH2 81880 FFAPL 57379 Delineation
- Page 735 and 736:
FOBT 60693 Useful For: Suggesting p
- Page 737 and 738:
FENTU 89655 testing often depend up
- Page 739 and 740:
FERR 8689 Clinical Information: Cli
- Page 741 and 742:
FECHK 60372 biochemical genetic tes
- Page 743 and 744:
FMBNY 30320 pregnancy, as well as b
- Page 745 and 746:
FGAKM 60722 in order to provide an
- Page 747 and 748:
FIBR 8482 FBC 80333 FGF23 88662 Fem
- Page 749 and 750:
FFIL4 90068 FIL 9232 The detected c
- Page 751 and 752:
FANT 82698 FBSH 82735 Laboratory Me
- Page 753 and 754:
9804 FLEC 9243 growth factor-bindin
- Page 755 and 756:
FFLRO 91795 FL 8641 impaired patien
- Page 757 and 758:
FFLUR 90091 17BFP 89739 ST. PAUL, M
- Page 759 and 760:
FSH 8670 folate deficiency states.
- Page 761 and 762:
FDP1 86207 antibodies interact with
- Page 763 and 764:
FOOD4 81872 3 3.50-17.4 Positive 4
- Page 765 and 766:
FOOD1 81868 sensitivity to inhalant
- Page 767 and 768:
FOOD3 81871 FRMH 82869 Clinical Ref
- Page 769 and 770:
9881 60694 Reporting limit determin
- Page 771 and 772:
FRANC 91552 other FMR1-related diso
- Page 773 and 774:
FFRED 91819 FFRBS 60476 Reference V
- Page 775 and 776:
FRUCT 81610 FROS 81164 GFDMS protei
- Page 777 and 778:
FSS 83121 FSC 83120 family member.
- Page 779 and 780:
FGEN 84389 FVAG 5184 after 30 days
- Page 781 and 782:
FFURO 91119 FUSM 82750 Furosemide (
- Page 783 and 784:
GABA 80826 cerebrospinal fluid may
- Page 785 and 786:
GDUR 89316 myocardium have also bee
- Page 787 and 788:
GALK 8628 associated with the nephr
- Page 789 and 790:
GALU 8765 GAL1P 80337 Galactose, Qu
- Page 791 and 792:
GALTP 80341 of GALT enzyme activity
- Page 793 and 794:
GALTK 84367 (galactose and galactos
- Page 795 and 796:
CBGT 8297 frequently observed mutat
- Page 797 and 798:
GCC 81981 Galectin-3 is a biomarker
- Page 799 and 800:
GGT 8677 interpretation and reporti
- Page 801 and 802:
GM1B 83189 Ganglioside Antibody Pan
- Page 803 and 804:
FGIP 90171 GAST 8512 1 0.35-0.69 Eq
- Page 805 and 806:
GBAMS 60711 GBAKM 60712 fasting per
- Page 807 and 808:
GELA 86326 diagnosis in at-risk pre
- Page 809 and 810:
GENPK 84695 GENT 81750 amyloidosis,
- Page 811 and 812:
GERB 82545 89713 Am Fam Physician 1
- Page 813 and 814:
GRAB 80628 GIAR 80231 3 3.50-17.4 P
- Page 815 and 816:
DGLDN 89031 2 0.70-3.49 Positive 3
- Page 817 and 818:
equires a jejunal biopsy demonstrat
- Page 819 and 820:
FGLIP 91097 GBM 8106 Disease Diagno
- Page 821 and 822:
GPI 9158 GLBF 8343 then rise again
- Page 823 and 824:
RGLUR 89847 GLSF 152 GLUR 8412 Gluc
- Page 825 and 826:
GD65C 84221 marker of predispositio
- Page 827 and 828:
FGLYA 57287 FGLMA 91742 sensitizati
- Page 829 and 830:
GMILK 82550 sensitivity to inhalant
- Page 831 and 832:
9810 9812 FGNRH 90165 GOOS 82714 Cl
- Page 833 and 834:
GRAM 8078 81990 LAGGT 8976 Referenc
- Page 835 and 836:
GRFR 82836 GRAS1 81706 Laboratory M
- Page 837 and 838:
GRAS3 81708 concentration of IgE an
- Page 839 and 840:
GNEM 82844 immunoglobulin E (IgE)-s
- Page 841 and 842:
GPEP 82623 2 0.70-3.49 Positive 3 3
- Page 843 and 844:
GRHMS 50037 proteins) followed by r
- Page 845 and 846:
9814 9815 ABO_M 9012 FGHBP 91958 an
- Page 847 and 848:
FGCU 57482 GGUM 82479 Patients with
- Page 849 and 850:
GUIN 82706 1 0.35-0.69 Equivocal 2
- Page 851 and 852:
HIBS 83261 HAKE 82348 Haemophilus i
- Page 853 and 854:
HALO 80339 4 17.5-49.9 Strongly pos
- Page 855 and 856:
HAPT 9168 Virus (SNV), which causes
- Page 857 and 858:
FHDLS 90186 FHE4 57164 proteins) fo
- Page 859 and 860:
ingestion, whereas MMA and DMA are
- Page 861 and 862:
HMSU 8633 HMSRU 60236 Reference val
- Page 863 and 864:
SHELA 84409 SHELP 84407 MERCURY 0-1
- Page 865 and 866:
SHELM 84408 is a convenient, noninv
- Page 867 and 868:
HELM 82749 disease, low-grade gastr
- Page 869 and 870:
HLLFH 34854 5434 molecular prognost
- Page 871 and 872:
HHEMO 81508 States are homozygous f
- Page 873 and 874:
A2F 83341 A1c (HbA1c) is a result o
- Page 875 and 876:
HBF 8269 usually easily identified
- Page 877 and 878:
SDEX 9180 Clinical Information: The
- Page 879 and 880:
HAPB 80297 stomatocytes, polychroma
- Page 881 and 882:
FIXMS 84209 HQ 9220 Hemophilia B, F
- Page 883 and 884:
FHPCF 91658 HIT 81904 preparations
- Page 885 and 886:
FHVP 90406 HAVM 8342 patients treat
- Page 887 and 888:
HAVAB 32110 result indicates immuni
- Page 889 and 890:
HBIS 209102 Core Antibody, IgM, Ser
- Page 891 and 892:
HBABT 87893 appear. It is detectabl
- Page 893 and 894:
HBAGP 86185 HBAG 9013 Instructions.
- Page 895 and 896:
QHBV 82416 serum (after it had beco
- Page 897 and 898:
HEAG 8311 remains detectable for se
- Page 899 and 900:
HCVPS 13009 Clinical References: 1.
- Page 901 and 902:
HCV 80190 "but >69,000,000 IU/mL" i
- Page 903 and 904:
HCVQU 83142 Interpretations: Quanti
- Page 905 and 906:
HCCAD 87858 HCVG 81618 Hepatitis C
- Page 907 and 908:
FHED 91850 FHEPD 91335 FHEVG 91222
- Page 909 and 910:
HEPS 200830 Reference Values: HEPAT
- Page 911 and 912:
FHER2 81954 Treat 1997 43:87-95. Mo
- Page 913 and 914:
81504 60198 tumors.(1) Specimens wi
- Page 915 and 916:
ACVK 89393 available to dimerize wi
- Page 917 and 918:
HHTM 89587 approximately 50% of dia
- Page 919 and 920:
HNPCC 17073 overall incidence of HH
- Page 921 and 922:
HP 83019 HSEP 81087 involvement of
- Page 923 and 924:
MHSV 87998 Clinical Information: He
- Page 925 and 926:
HSV 84422 Coombs RW, Benedetti J, e
- Page 927 and 928:
FHHV6 91311 FHV6D 57484 FHP6 80419
- Page 929 and 930:
FHART 57462 FHEXA 91442 HEXAI 82397
- Page 931 and 932:
NAGT 8776 Clinical References: 1. T
- Page 933 and 934:
y 4 years. Tay-Sachs disease is an
- Page 935 and 936:
NAGR 82943 TOTAL Reference values h
- Page 937 and 938:
FHSTW 57368 HMAX 5338 HIS 80944 Age
- Page 939 and 940:
HBRP 60213 antibodies is presumptiv
- Page 941 and 942:
RHIV 84455 Reference Range: Negativ
- Page 943 and 944:
HV1CM 60357 verified by submitting
- Page 945 and 946:
GHIV 82340 HIV, as well as nonviabl
- Page 947 and 948:
PHIV 88635 genotypic mutations pres
- Page 949 and 950:
limit of this assay. A "Detected" r
- Page 951 and 952:
HIVQU 81958 susceptibility to the s
- Page 953 and 954:
WBAR 23878 suspected or repeat HIV
- Page 955 and 956:
HIV2 86702 patients reside. A negat
- Page 957 and 958:
FHLAA 91498 FHLAB 91499 FHLA 91833
- Page 959 and 960:
HL15O 60348 HLA57 89346 An interpre
- Page 961 and 962:
LY27B 9648 HMBSS 61216 histocompati
- Page 963 and 964:
HCYSP 80379 in 350,000 live births.
- Page 965 and 966:
HVA 9253 HVAR 60275 Interpretation:
- Page 967 and 968:
HBV 82551 Class IgE kU/L Interpreta
- Page 969 and 970:
HORS 82874 immune response to aller
- Page 971 and 972:
HSPR 82134 4 17.5-49.9 Strongly pos
- Page 973 and 974:
DP 82904 Useful For: Testing for Ig
- Page 975 and 976:
HDG 82906 HDHS 82903 Clinical Refer
- Page 977 and 978:
FHUAB 57246 FHAMA 57151 THCG 80678
- Page 979 and 980:
HHV6V 89888 HHV8 81971 Clinical Ref
- Page 981 and 982:
80172 significance" (ASCUS) on Pap
- Page 983 and 984:
needles. Two diseases are known to
- Page 985 and 986:
FHIME 91376 Reference Values: Negat
- Page 987 and 988:
SEROTYPE 3 (3) mcg/mL SEROTYPE 9 (9
- Page 989 and 990:
FHYCO 90110 FHYCD 91637 HYMP 9736 N
- Page 991 and 992:
FVIST 90121 HMDP 89220 Adults 8:00
- Page 993 and 994:
HYOX 86213 CD19+ CD27+ IgM+ IgD+ 1.
- Page 995 and 996:
SAL 8768 Aspergillus fumigatus #6 A
- Page 997 and 998:
HIF2A 61681 61207 Interpretation: U
- Page 999 and 1000:
FIFAF 91181 FSAGA 90047 X-linked di
- Page 1001 and 1002:
FIGBP 57131 FIGF2 80758 16- or =18
- Page 1003 and 1004:
CASF 8271 16- or =18 years: 341-894
- Page 1005 and 1006:
IMPR 8126 CMPD. These translocation
- Page 1007 and 1008:
FICP 91173 FISP 91624 C3, NEPH REFE
- Page 1009 and 1010:
FIMM 91507 IGA 8157 urine protein e
- Page 1011 and 1012:
IGE 8159 FLCP 84190 features, respo
- Page 1013 and 1014:
BCGR 83123 BCGBM 31141 9-
- Page 1015 and 1016:
IGM 8158 IGGS4 84250 Immunoglobulin
- Page 1017 and 1018:
IMMG 8156 KAPPA TOTAL LIGHT CHAIN
- Page 1019 and 1020:
MONOS 9081 IBDP 81443 Salt Lake Cit
- Page 1021 and 1022:
SFLA 8169 SFLB 8175 Influenza Virus
- Page 1023 and 1024:
FLUAB 60551 Influenza Virus Type A
- Page 1025 and 1026:
INHAB 86336 INHA 81049 > or =16 yea
- Page 1027 and 1028:
phase decline in FSH levels. Inhibi
- Page 1029 and 1030:
INAB 8666 6 > or =100 Strongly posi
- Page 1031 and 1032:
IGFP 83357 Clinical References: Thr
- Page 1033 and 1034:
15 years 236-1,060 153 16 years 227
- Page 1035 and 1036:
80 years 53-162 Reference values ha
- Page 1037 and 1038:
acromegaly or gigantism in individu
- Page 1039 and 1040:
IGFB3 83300 46-50 years 91-246 51-5
- Page 1041 and 1042:
FINTE 90483 31-35 years: 3.5-7.0 mc
- Page 1043 and 1044:
OIL28 61701 3-fold greater rates of
- Page 1045 and 1046:
FIL2R 90021 FINL6 91979 FIL8 91654
- Page 1047 and 1048:
IODU 8639 IOD 81574 ICRU 60440 Corr
- Page 1049 and 1050:
FEU 8571 FET 8350 Reference Values:
- Page 1051 and 1052:
FEUR 88970 FECRU 60764 A: Hemochrom
- Page 1053 and 1054:
BTITH 8972 IHDI 82773 Clinical Info
- Page 1055 and 1056:
ITDT 82775 concentration of IgE ant
- Page 1057 and 1058:
ISPG 82768 ITCON 81247 newborn scre
- Page 1059 and 1060:
JMACK 82819 0 Negative 1 0.35-0.69
- Page 1061 and 1062:
JAK2B 88715 chronic myelogenous leu
- Page 1063 and 1064:
JAK2V 31156 JCEDR 82865 Buser AS, e
- Page 1065 and 1066:
FJCV 91827 81107 Reference Values:
- Page 1067 and 1068:
JOHN 82900 symptoms, Raynaudâ€s
- Page 1069 and 1070:
FKAL 88540 FKAN 90069 Results are e
- Page 1071 and 1072:
89027 KIDBN 82619 Reference Values:
- Page 1073 and 1074:
KITAS 88802 Interpretation: The int
- Page 1075 and 1076:
88957 88955 dysphagia. Clin Cancer
- Page 1077 and 1078:
FXYM 86374 immune response to aller
- Page 1079 and 1080:
GALCK 60695 between 3 to 6 months o
- Page 1081 and 1082:
FLACO 57111 LD_I 8679 receptor-targ
- Page 1083 and 1084:
Interpretation: Marked elevations i
- Page 1085 and 1086:
LAMQ 82682 LAMB 82699 Positive 4 >
- Page 1087 and 1088:
LBC 60450 utilizing the platelet ch
- Page 1089 and 1090:
LANG 82349 FLTX 57118 Langust (Lobs
- Page 1091 and 1092:
LEADO 300115 recognized as a risk f
- Page 1093 and 1094:
PBU 8600 produced for nondomestic u
- Page 1095 and 1096:
PBHA 8495 PBNA 89857 standards for
- Page 1097 and 1098:
LCATD 83253 C, Buchet JP, Leroyer A
- Page 1099 and 1100:
LEGI 8204 estimated to be responsib
- Page 1101 and 1102:
LEGRP 89564 LEIS 86219 elevated as
- Page 1103 and 1104:
LEPD 82849 antibodies interact with
- Page 1105 and 1106:
LETT 82805 Useful For: As an aid in
- Page 1107 and 1108:
LLPT 19499 LAD1 81155 Leukemia/Lymp
- Page 1109 and 1110:
LID 8382 FLIMB 91635 LIME 82360 0.2
- Page 1111 and 1112:
LIND 82862 LINS 86311 Linden, IgE C
- Page 1113 and 1114:
FLIPR 90347 LPS 8328 BFLAC 34622 LP
- Page 1115 and 1116:
LPAWS 89005 Low HDL: or =60 mg/dL
- Page 1117 and 1118:
FLPA2 57353 LMPP 83673 would be >5
- Page 1119 and 1120:
FLIS 90717 TRIGLYCERIDES Normal: o
- Page 1121 and 1122:
LKM 80387 LOB 82744 Product Monogra
- Page 1123 and 1124:
FLCA 60619 8-Hydroxyloxapine: Refer
- Page 1125 and 1126:
LUPPR 83092 LH 8663 Test Performed
- Page 1127 and 1128:
9861 9956 PBORR 80574 Follicular: 2
- Page 1129 and 1130:
FBBIA 91898 FBBAB 91309 Serology is
- Page 1131 and 1132:
CLYWB 83857 FBBC6 91899 after onset
- Page 1133 and 1134:
CLYME 83856 diagnosis. Useful For:
- Page 1135 and 1136:
LPAGF 60592 Maximum proliferation o
- Page 1137 and 1138:
LPMGF 60591 recognition. Chem Rev 1
- Page 1139 and 1140:
FLCM 90042 LSDU 81743 birth to old
- Page 1141 and 1142:
deficiency of sphingomyelinase, whi
- Page 1143 and 1144:
LYZKM 60720 amyloidosis, with renal
- Page 1145 and 1146:
FMNUT 91661 MACE 82492 Reference Va
- Page 1147 and 1148:
MCRPL 87843 MGFT 60030 6 > or =100
- Page 1149 and 1150:
MGF 81345 MGRU 60245 diuretics) enh
- Page 1151 and 1152:
MGCRU 60244 Useful For: Magnesium l
- Page 1153 and 1154:
MAL 9240 MAAN 82396 Plasmodium spec
- Page 1155 and 1156:
MAND 82352 sensitization to particu
- Page 1157 and 1158:
isocitrate dehydrogenase. It circul
- Page 1159 and 1160:
MNS 8413 MNCRU 60027 Manganese, Pla
- Page 1161 and 1162:
MBL 81051 1 0.35-0.69 Equivocal 2 0
- Page 1163 and 1164:
MARE 82141 changes in behavior, dif
- Page 1165 and 1166:
9828 MCC 88636 9230 6 > or =100 Str
- Page 1167 and 1168:
MFOX 82914 immune response to aller
- Page 1169 and 1170:
ME2KM 89285 with a clinical diagnos
- Page 1171 and 1172:
MCADK 83934 acylcarnitines (ACRN/82
- Page 1173 and 1174:
MELAI 82724 FMELT 57120 MELN Melale
- Page 1175 and 1176:
This assay was developed and its pe
- Page 1177 and 1178:
FMCPC 57437 Reference Range: Negati
- Page 1179 and 1180:
Diagnosis of infections of the cent
- Page 1181 and 1182:
levels found in CSF, passive transf
- Page 1183 and 1184:
symptoms. The presence of mumps IgG
- Page 1185 and 1186:
MEPHS 83778 FMERC 91120 HGOM 82755
- Page 1187 and 1188:
HGHAR 8498 in 3 ways: -Hg(+2) is re
- Page 1189 and 1190:
MESQ 82776 METAF 83006 Mesquite, Ig
- Page 1191 and 1192:
PMET 81609 13-17 years: 57-286 mcg/
- Page 1193 and 1194:
nonepisodic hypertension Interpreta
- Page 1195 and 1196:
MTDNS 83131 undergoing treatment wi
- Page 1197 and 1198:
METR 9322 MEVP 84159 sulfhemoglobin
- Page 1199 and 1200:
FMMD 57307 MMAAF 81921 Methsuximide
- Page 1201 and 1202:
MMAS 80289 Useful For: Evaluating c
- Page 1203 and 1204:
MAHKM 89135 Clinical Information: M
- Page 1205 and 1206:
MHDKM 61098 Carrier screening in ca
- Page 1207 and 1208:
FMI2 57186 RMA 81260 been shown for
- Page 1209 and 1210:
MPSF 82515 whether this needs to be
- Page 1211 and 1212:
MTBS 81507 leucovorin (LV). These f
- Page 1213 and 1214:
PMLK 82827 0 Negative 1 0.35-0.69 E
- Page 1215 and 1216:
AMA 8176 Clinical Information: Here
- Page 1217 and 1218:
MLH1H 87978 Usual therapeutic doses
- Page 1219 and 1220:
MLHKM 83002 hereditary defective mi
- Page 1221 and 1222:
MCDMS 89830 and endometrial cancer.
- Page 1223 and 1224:
MOLD1 81878 MINT 61696 MOLU 89271 M
- Page 1225 and 1226:
MOLPS 89270 appetite, tachycardia,
- Page 1227 and 1228:
FMNM 91829 124 healthy adults by Ma
- Page 1229 and 1230:
identification of a monoclonal band
- Page 1231 and 1232:
MPSU 8823 considered an adequate sc
- Page 1233 and 1234:
MDCG 86880 MORP 83132 SPSM 9184 Mon
- Page 1235 and 1236:
MOTH 82738 FMOT 90157 Chapter 53, P
- Page 1237 and 1238:
MOUS 82707 MOSP 82792 Mouse Epithel
- Page 1239 and 1240:
9832 MPLB 89776 0 Negative 1 0.35-0
- Page 1241 and 1242:
MSH2M 83016 mutation is identified
- Page 1243 and 1244:
MSH6M 83723 instability and immunoh
- Page 1245 and 1246:
9831 MCIV 85321 MPSSC 84464 Mucicar
- Page 1247 and 1248:
MPSQN 81473 dysostosis multiplex, f
- Page 1249 and 1250:
MUG 82683 allergic reactions to ins
- Page 1251 and 1252:
MENKM 81082 MENMS 80573 Multiple En
- Page 1253 and 1254:
FMUMM 91456 CMUMP 81435 0-4 months:
- Page 1255 and 1256:
MMPM 80977 (meningitis/encephalitis
- Page 1257 and 1258:
FMUSK 91445 MSTD 82801 6 > or =100
- Page 1259 and 1260:
FHIST 90018 FHSAG 90017 FHSU 90019
- Page 1261 and 1262:
MGEP 83371 the Lambert-Eaton myasth
- Page 1263 and 1264:
MGLES 83369 Negative AChR GANGLIONI
- Page 1265 and 1266:
CTBBL 82443 FMC12 91558 Useful For:
- Page 1267 and 1268:
MTBPZ 56099 QTBG 83896 Interpretati
- Page 1269 and 1270:
MGRP 60755 MHRP 60756 overimmunosup
- Page 1271 and 1272:
FMPAB 90055 MPC 80422 FMPD 91429 FM
- Page 1273 and 1274:
FMDS 84387 FMFC 60405 Test Performe
- Page 1275 and 1276:
MYH 84304 MCA 9746 MYH Gene Analysi
- Page 1277 and 1278:
MYOS 9035 MYOU 9274 Single titers o
- Page 1279 and 1280:
FMYO 91544 FCHOP 84456 Reference Va
- Page 1281 and 1282:
NAT2O 60345 hepatitis, peripheral n
- Page 1283 and 1284:
FNTPX 57308 mast-cell activity, suc
- Page 1285 and 1286:
NARC 82026 NKCP 28562 Test Performe
- Page 1287 and 1288:
6-11 months: 31-56%* 12-23 months:
- Page 1289 and 1290:
MGRNA 61646 Test Performed By Medto
- Page 1291 and 1292:
FNMEN 91669 FNEOM 90288 (eg, cultur
- Page 1293 and 1294:
NEURF 88846 FNMYC 87862 Reference V
- Page 1295 and 1296:
Clinical Information: Paraneoplasti
- Page 1297 and 1298:
FNEA 57115 NEEVP 84162 utilizing PN
- Page 1299 and 1300:
NMOER 60796 for neuromyelitis optic
- Page 1301 and 1302:
NSESF 81796 Clinical References: 1.
- Page 1303 and 1304:
FNTSM 91940 DISCLAIMER required by
- Page 1305 and 1306:
FNIAC 91379 NIU 8626 Reference Valu
- Page 1307 and 1308:
NIS 8622 NICRU 60442 Environmental
- Page 1309 and 1310:
NICOU 82510 Interpretation: Serum n
- Page 1311 and 1312:
NPDKM 61116 NPD mutations in indivi
- Page 1313 and 1314:
NPCKM 83118 NPCMS 89015 abnormal me
- Page 1315 and 1316:
NITU 8586 NMDCS 61516 muscle protei
- Page 1317 and 1318:
FNMR 91959 imaging (MRI) of pelvis,
- Page 1319 and 1320:
SSF1 87294 NSIP 31769 NRDZ 501030 N
- Page 1321 and 1322:
NEREG 31767 NORT 81858 Cypress, CA
- Page 1323 and 1324:
54 years: 10-67 pg/mL 55 years: 10-
- Page 1325 and 1326:
NPM1 89292 should be considered. Wh
- Page 1327 and 1328:
OAK 82673 immune response to allerg
- Page 1329 and 1330:
OCC2 81870 4 17.5-49.9 Strongly pos
- Page 1331 and 1332:
OLIG 8017 OLIGO 84340 402 W. County
- Page 1333 and 1334:
OLIVF 86306 4 17.5-49.9 Strongly po
- Page 1335 and 1336:
OPATU 8473 semisynthetic narcotic d
- Page 1337 and 1338:
OPRMO 60352 haplotype-based approac
- Page 1339 and 1340:
OREG 82496 caused by the release of
- Page 1341 and 1342:
IDENT 9221 ANIDE 8114 terms only. W
- Page 1343 and 1344:
UOSMB 9257 UOSMF 9258 hematuria, re
- Page 1345 and 1346:
FRAG 9064 OSCAL 80579 dehydration,
- Page 1347 and 1348:
OVAL 82826 Low High Premenopausal
- Page 1349 and 1350:
DOXA 61644 proteins) followed by re
- Page 1351 and 1352:
OXU 8669 of oxalate can be increase
- Page 1353 and 1354:
OXYCS 83654 P50 9110 Oxycodone ng/m
- Page 1355 and 1356:
SQUI 82821 This test was developed
- Page 1357 and 1358:
PAIN 86328 drugs of abuse. Opiate c
- Page 1359 and 1360:
PAPN 82383 Useful For: Detection of
- Page 1361 and 1362:
PARAV 80421 immunoglobulin E (IgE)-
- Page 1363 and 1364:
PNEOE 80013 NEURONAL AND MUSCLE CYT
- Page 1365 and 1366:
PARID 9202 OAP 9216 reported as "un
- Page 1367 and 1368:
PTH2P 28380 levels, who have either
- Page 1369 and 1370:
PTHFN 61526 Females 0-11 months: no
- Page 1371 and 1372:
PCAB 83728 when tumors secrete PTHr
- Page 1373 and 1374:
POFF 82549 PARO 83731 Laboratory Me
- Page 1375 and 1376:
FPDF 91577 FPDM 91576 PARV 84325 Cl
- Page 1377 and 1378:
PARVP 86337 host.(2,3) Infection wi
- Page 1379 and 1380:
PCBP 80587 88905 indicated for use
- Page 1381 and 1382:
88958 89672 PDGFRA, Mutation Analys
- Page 1383 and 1384:
FPEAG 91999 FPEA4 91981 Interpretat
- Page 1385 and 1386:
PEC 82880 bronchospasm) in infants
- Page 1387 and 1388:
PAS3 83345 PAS8 83347 6 > or =100 S
- Page 1389 and 1390:
PENG 80134 immunoglobulin E (IgE)-s
- Page 1391 and 1392:
FFTAL 90099 PENTS 8239 Clinical Inf
- Page 1393 and 1394:
9840 9839 PNPC 5341 ACASM 83632 Not
- Page 1395 and 1396:
PERS 82353 UPH24 84047 Persimmon, I
- Page 1397 and 1398:
FPHAI 91629 PCPUG 9788 PCPMC 89069
- Page 1399 and 1400:
FPGT 91757 FPFUZ 91755 FPHIV 91756
- Page 1401 and 1402:
FPEMA 90102 PHYF 6794 Conversion Fo
- Page 1403 and 1404:
P_PB 8643 PNY 8604 Clinical Referen
- Page 1405 and 1406:
FPHAB 57371 FPHOS 57310 ACLIP 86179
- Page 1407 and 1408:
CLPMG 82976 2006;4: 295-306 5. Font
- Page 1409 and 1410:
GCLIP 80993 15.0-39.9 APL (weakly p
- Page 1411 and 1412:
PPL 8296 cross-linking beta 2 GP1 m
- Page 1413 and 1414:
PMMIL 89656 Interpretation: Normal
- Page 1415 and 1416:
PHBF 8029 RPOU 84007 POU 8526 West
- Page 1417 and 1418:
PANH 81156 6 >or =100 Strongly posi
- Page 1419 and 1420:
SPB 8892 PDRP 82796 Laboratory Meth
- Page 1421 and 1422:
PINE 82381 PNAP 82815 Pine Nut, IgE
- Page 1423 and 1424:
PIPU 81248 Clinical Information: Pi
- Page 1425 and 1426:
PIOR 82851 FPIV 91493 Pityrosporum
- Page 1427 and 1428:
PCPRO 61654 therefore, is an advers
- Page 1429 and 1430:
FPCPD 83358 PLHBB 9096 Plasma Cell
- Page 1431 and 1432:
FPACT 91760 PSGN 9079 Hemostasis an
- Page 1433 and 1434:
PLUM 82809 drugs Interpretation: Ef
- Page 1435 and 1436:
FPMP 91590 PMS2S 61173 Clinical Ref
- Page 1437 and 1438:
FPNEU 91656 PNRP 81698 Long-range P
- Page 1439 and 1440:
POLIO 80420 FPOLS 91469 GAAMS 89898
- Page 1441 and 1442:
FPLWH 91991 POPSD 82632 Hirschhorn
- Page 1443 and 1444:
PORK 82700 PBGDW 31894 Clinical Ref
- Page 1445 and 1446:
PBGU 82068 Useful For: Confirmation
- Page 1447 and 1448:
PEE 88886 a detailed interpretation
- Page 1449 and 1450:
FQPPS 81652 UROPORPHYRIN < or =2 mc
- Page 1451 and 1452:
counseling of the patient regarding
- Page 1453 and 1454:
oth coproporphyrin and PBG excretio
- Page 1455 and 1456:
FPOS 91997 POSA 89591 with lesser i
- Page 1457 and 1458:
NAK 8468 dehydrogenase (LCHAD) defi
- Page 1459 and 1460:
KBF 8028 KF 8375 Potassium, Body Fl
- Page 1461 and 1462:
KUR 8527 FPOTW 92002 with rapid K i
- Page 1463 and 1464:
PALB 9005 FPRGA 57110 17PRN 88646 R
- Page 1465 and 1466:
10-12 years: 19-220 ng/dL 13-15 yea
- Page 1467 and 1468:
FPAP2 91198 Reference Values: CHILD
- Page 1469 and 1470:
Units: ng/dL Age Range Premature (2
- Page 1471 and 1472:
PHESP 9021 products (ie, blood tran
- Page 1473 and 1474:
PTRE 82784 adjustment based on bloo
- Page 1475 and 1476:
detectable within 2 to 4 hours afte
- Page 1477 and 1478:
FPROG 90287 GRNMS 89188 *Lippe BM,
- Page 1479 and 1480:
PRLPM 84462 Clinical Information: P
- Page 1481 and 1482:
PROCT 83097 PHD2 61683 prolactin le
- Page 1483 and 1484:
FIBDD 57459 FPLAC 91783 FPMET 91564
- Page 1485 and 1486:
FPROP 90362 FPPOX 57140 FPRSG 57149
- Page 1487 and 1488:
FPSAP 91775 PSA 9284 Prostate Speci
- Page 1489 and 1490:
PSAFT 81944 > or =80 < or =7.2 Fema
- Page 1491 and 1492:
PROT 82139 CFX 9339 of pretreatment
- Page 1493 and 1494:
S_FX 80775 Clinical Information: Ph
- Page 1495 and 1496:
factors Va and VIIIa. In addition,
- Page 1497 and 1498:
TPBF 8420 RPTU 85681 accurately ass
- Page 1499 and 1500:
PTU 8261 PR3 82965 Interpretation:
- Page 1501 and 1502:
PT 9236 C, protein S, or antithromb
- Page 1503 and 1504:
PPFE 8739 Noncomplexed (free) proto
- Page 1505 and 1506:
PCHES 8518 (0%-20%) and usually low
- Page 1507 and 1508:
FPTH 90182 PT1K 89464 9236 ABRAHAM
- Page 1509 and 1510:
PTP22 89315 PTPN11 are missense mut
- Page 1511 and 1512:
PUSE 82362 Clinical References: 1.
- Page 1513 and 1514:
FPYTH 91667 FPYD 90281 PLP 60295 >
- Page 1515 and 1516:
PK 8659 of PDHC deficiency is a def
- Page 1517 and 1518:
QFP 83149 Reference Values: 0.08-0.
- Page 1519 and 1520:
ovarian granulosa cells and testicu
- Page 1521 and 1522:
FQUET 91727 QUIN 8302 REPII 82782 C
- Page 1523 and 1524:
RSER 82544 0 Negative 1 0.35-0.69 E
- Page 1525 and 1526:
RASE 82366 Reference Values: Report
- Page 1527 and 1528:
RASP 86305 Interpretation: Treatmen
- Page 1529 and 1530:
RTUP 82794 proteins) followed by re
- Page 1531 and 1532:
SORR 82737 Clinical Information: Cl
- Page 1533 and 1534:
UREDF 83255 4986 1 0.35-0.69 Equivo
- Page 1535 and 1536:
88501 PRA 8060 Clinical References:
- Page 1537 and 1538:
SRSV 8301 RSVAN 110300 and Thrombos
- Page 1539 and 1540:
RTIC 9108 FREB 90331 RBP24 81783 Mu
- Page 1541 and 1542:
FRFSF 57516 RHUT 9060 RHNI 82856 Rh
- Page 1543 and 1544:
RIBAV 60536 VITB2 61637 Ribavirin,
- Page 1545 and 1546:
FRCBP 57342 Useful For: Testing for
- Page 1547 and 1548:
RNAP 83397 myeloproliferative disea
- Page 1549 and 1550:
ROTA 8886 MARS 82701 Test Performed
- Page 1551 and 1552:
RB 8172 ROSC 80262 1 0.35-0.69 Equi
- Page 1553 and 1554:
ROC 5194 ROM Reference Values: Nega
- Page 1555 and 1556:
RYEG 82908 proteins) followed by re
- Page 1557 and 1558:
F100B 57349 AASCA 83022 6 > or =100
- Page 1559 and 1560:
SALC 8480 SALM 82754 Salicylate, Se
- Page 1561 and 1562:
SARD 82818 sensitization to particu
- Page 1563 and 1564:
SCALS 82259 SHUR 60451 Scallop, IgE
- Page 1565 and 1566:
SCL70 80178 OXK 8148 SEAFP 31770 Sc
- Page 1567 and 1568:
SECOS 8243 FSEC 90173 sensitization
- Page 1569 and 1570:
SEUR 60077 Teratogenic effects are
- Page 1571 and 1572:
FER 81641 Clinical Information: Sel
- Page 1573 and 1574:
FSMN 91449 SEPTK 61101 clinical man
- Page 1575 and 1576:
screening has a higher detection ra
- Page 1577 and 1578:
directly into the amniotic fluid ca
- Page 1579 and 1580:
HTR2O 60338 genotype.(4) - For the
- Page 1581 and 1582:
HTTO 60339 (SSRIs) Evaluating patie
- Page 1583 and 1584:
SERWB 84373 elevated in nearly all
- Page 1585 and 1586:
can release 5-HT from EC-cells. Onc
- Page 1587 and 1588:
SESA 82728 SHBG 9285 Sesame Seed, I
- Page 1589 and 1590:
FSRY 88537 Reference ranges for pre
- Page 1591 and 1592:
Class IgE kU/L Interpretation 0 Neg
- Page 1593 and 1594:
SCADK 83947 sensitization and clini
- Page 1595 and 1596: FSHOX 57127 FSHRP 91650 SHRI 82677
- Page 1597 and 1598: BIR 82674 and to define the allerge
- Page 1599 and 1600: SIRO 81768 SM 81358 Following a sin
- Page 1601 and 1602: FSMS 88534 SMA 6284 Quantitative re
- Page 1603 and 1604: NAFT 60032 NABF 8039 NAF 8374 2007
- Page 1605 and 1606: NACCL 81692 NAU 8525 Clinical Refer
- Page 1607 and 1608: FSLA 91610 STFR 84283 Laboratory Me
- Page 1609 and 1610: FSOMA 90172 associated with statins
- Page 1611 and 1612: SOY 82886 SPAG 8980 Soybean, IgE Le
- Page 1613 and 1614: SAAS 89882 SAAI 89883 questioned. T
- Page 1615 and 1616: SPIN 86312 FSMAC U/g of cellular pr
- Page 1617 and 1618: FSCA2 91586 FSCA3 91587 FSCA6 91588
- Page 1619 and 1620: SFGP 83679 SPRU 82394 Clinical Refe
- Page 1621 and 1622: SQUID 82631 FSRP 57187 Clinical Ref
- Page 1623 and 1624: SSB 81359 with childhood LE, neonat
- Page 1625 and 1626: ST2S 61723 signs. The severity of i
- Page 1627 and 1628: FSTS 88539 Clinical Information: Cl
- Page 1629 and 1630: INSEC 31765 STBY 82676 Clinical Ref
- Page 1631 and 1632: SPNC 89971 SPNEU 83150 Although the
- Page 1633 and 1634: concentrations of IgG antibodies to
- Page 1635 and 1636: FSTRP 90440 FSTSC 91984 STR 8746 J
- Page 1637 and 1638: FSAI 57313 STCH 9928 FSTYR 91094 Ge
- Page 1639 and 1640: SDHSP 89550 Useful For: Evaluation
- Page 1641 and 1642: SDHKM 89554 is higher than that of
- Page 1643 and 1644: SDHSB 89551 When such correlations
- Page 1645: SDHSD 89553 this subtype. SDHD show
- Page 1649 and 1650: SFZ 8238 identify allergens which m
- Page 1651 and 1652: SUNFS 82813 SUNF 82615 IgG < 1500 >
- Page 1653 and 1654: adiopaque stone, for whom stone ana
- Page 1655 and 1656: oral potassium citrate will raise t
- Page 1657 and 1658: SNS 82594 Supplemental Newborn Scre
- Page 1659 and 1660: 5581 SGUM 82483 Note: This test is
- Page 1661 and 1662: VERG 82909 SWORD 82346 Company, 200
- Page 1663 and 1664: 83361 SS18-SSX2 fusion. Unfortunate
- Page 1665 and 1666: SGSU 81035 the risks of systemic ad
- Page 1667 and 1668: SYPGN 32184 of reverse screening re
- Page 1669 and 1670: TBNY 82589 results of the first tre
- Page 1671 and 1672: TCIPF 60590 3-5 months: 170-830 cel
- Page 1673 and 1674: ABSOLUTE COUNTS CD45 Lymph Count, F
- Page 1675 and 1676: 0-2 months: 6-32%* 3-5 months: 11-4
- Page 1677 and 1678: TCMPF 60588 T- and B-Cell Quantitat
- Page 1679 and 1680: TBBS 9336 2-5 years: 700-2,200 cell
- Page 1681 and 1682: CD45 Lymph Count, Flow 0-17 years:
- Page 1683 and 1684: FRTLP 89040 89041 Clinical Referenc
- Page 1685 and 1686: during the process of TCR rearrange
- Page 1687 and 1688: TCGR 83122 > or =55 years: >78 copi
- Page 1689 and 1690: TCP 89319 Useful For: Determining w
- Page 1691 and 1692: CD8+CD45RO+ memory T cells 2-26% of
- Page 1693 and 1694: Foxp3+CD4+CD25+ T cells and provide
- Page 1695 and 1696: FRT3P 600915 FRT3 9404 fluctuations
- Page 1697 and 1698:
TUP 81792 Interpretation: Triiodoth
- Page 1699 and 1700:
T4TF 8684 T4 8724 salicylates. Inte
- Page 1701 and 1702:
TARR 82486 HEXMS 89278 Tarragon, Ig
- Page 1703 and 1704:
TSD 82588 Interpretation: An interp
- Page 1705 and 1706:
FFTEM 80763 TTBS 80065 TEST PERFORM
- Page 1707 and 1708:
esponse. Bioavailable (TTBS/80065):
- Page 1709 and 1710:
in SHBG (#9285 "Sex Hormone Binding
- Page 1711 and 1712:
TTFB 83686 variable. Tanner stage V
- Page 1713 and 1714:
Reference Values: TESTOSTERONE, TOT
- Page 1715 and 1716:
associated with increased luteinizi
- Page 1717 and 1718:
FFTEN 57102 THCU 8898 Reference Val
- Page 1719 and 1720:
TGF1 89459 beta R-I, propagating th
- Page 1721 and 1722:
TGF2 89461 (TAAD), which involves c
- Page 1723 and 1724:
THEVP 84158 TLU 8603 Thalassemia an
- Page 1725 and 1726:
FFTHC 90094 THEO 8661 1 gram. Usefu
- Page 1727 and 1728:
TDP 85753 of sera from patients wit
- Page 1729 and 1730:
83343 High Risk HPV DNA Detection:
- Page 1731 and 1732:
83342 with HPV effect -High-grade s
- Page 1733 and 1734:
TPMT 80291 Synonym(s): Pentothal Hy
- Page 1735 and 1736:
FFTAT 91200 THRMP 83093 The thrombi
- Page 1737 and 1738:
RTEP 89507 allergic reactions to in
- Page 1739 and 1740:
TAP 89506 1 month-17 years: 170.0-1
- Page 1741 and 1742:
12-23 months: 2,100-6,200 cells/mcL
- Page 1743 and 1744:
TGAB 84382 Thyroglobulin Antibody,
- Page 1745 and 1746:
HTG1 83069 This cutoff has been val
- Page 1747 and 1748:
STSH 8939 in Special Instructions.
- Page 1749 and 1750:
TPO 81765 associated with neonatal
- Page 1751 and 1752:
TBGI 9263 TBPE 8838 relevant in wom
- Page 1753 and 1754:
FFTIK 91818 TICKS 83265 Useful For:
- Page 1755 and 1756:
FFTLA 91951 TIMG 82891 Reference Va
- Page 1757 and 1758:
TTGA 82587 Genetic susceptibility i
- Page 1759 and 1760:
TTGG 83660 Tissue Transglutaminase
- Page 1761 and 1762:
TIS 89367 Clinical Information: Tit
- Page 1763 and 1764:
TOBAC 82620 creatinine in a patient
- Page 1765 and 1766:
TOBT 81594 FHIPP 91121 FFTLB 91141
- Page 1767 and 1768:
TOPI 81546 Reference Values: Class
- Page 1769 and 1770:
should be drawn and tested. Rubella
- Page 1771 and 1772:
FFTIH 91594 Negative CMV IgM result
- Page 1773 and 1774:
TOXOG 8267 amniotic fluid specimens
- Page 1775 and 1776:
PTOX 81795 FFTXG 91211 FFTXM INTERP
- Page 1777 and 1778:
FFTRA 91693 TNFN 300205 Company, 20
- Page 1779 and 1780:
TACIF 84388 Transmembrane Activator
- Page 1781 and 1782:
FFTRC 91091 TRZ 9624 Useful For: Id
- Page 1783 and 1784:
TREE3 81704 sensitivity to inhalant
- Page 1785 and 1786:
FFTPG 57315 FHAL 90119 STRIC 9017 F
- Page 1787 and 1788:
TRVI 82853 Trichloroethylene, Blood
- Page 1789 and 1790:
TRICY 500509 bronchospasm) in infan
- Page 1791 and 1792:
TGLBF 61647 TRIG 8316 DOXEPIN AND N
- Page 1793 and 1794:
FFTRM 90115 TMP 80146 SURM 80463 5
- Page 1795 and 1796:
FFTRP 91774 inherited neurodegenera
- Page 1797 and 1798:
WHIPB 87974 bacilli. Culture of Whi
- Page 1799 and 1800:
CHAG 86159 bronchospasm) in infants
- Page 1801 and 1802:
TRYPT 81608 TRYPP 82955 measurable
- Page 1803 and 1804:
RTRPP 88546 Inborn Errors of Metabo
- Page 1805 and 1806:
TURKF 82824 proteins) followed by r
- Page 1807 and 1808:
FFTUR 92005 TRYPI 82848 6 > or =100
- Page 1809 and 1810:
UBEKM 89920 UGTK Reference Values:
- Page 1811 and 1812:
UGT2 89611 II; CN type I does not r
- Page 1813 and 1814:
UGTI 89397 UGTIO 60349 UDP-Glucuron
- Page 1815 and 1816:
U1A1O 60343 when predicting the UGT
- Page 1817 and 1818:
FURA 90316 Clinical Information: Un
- Page 1819 and 1820:
FURBF 57373 RURCU 89846 urealyticum
- Page 1821 and 1822:
URCU 8529 RUA 9308 > or =16 years:
- Page 1823 and 1824:
UMIC 9316 Reference Values: Descrip
- Page 1825 and 1826:
UPGD 8599 Reference Values: 1.0-3.0
- Page 1827 and 1828:
60714 tumor suppressor gene). The U
- Page 1829 and 1830:
FVPA 81771 patients with uveal mela
- Page 1831 and 1832:
VUR 89680 VS 83396 Useful For: Dete
- Page 1833 and 1834:
VANPK 80141 VANCR 81749 Interpretat
- Page 1835 and 1836:
VRERP 84406 VANIL 82621 Therapy, Ma
- Page 1837 and 1838:
VMA 9454 VMAR 60274 HVA
- Page 1839 and 1840:
FVZVS 91685 SVZP 84424 are at risk
- Page 1841 and 1842:
LVZV 81241 FVEGF 91765 VIP 8150 Neg
- Page 1843 and 1844:
VENLA 83732 Clinical Information: C
- Page 1845 and 1846:
VLCKM 60037 VIBC 89658 Clinical Ref
- Page 1847 and 1848:
VISCS 8168 VAE 60299 diagnosis by i
- Page 1849 and 1850:
VITA 60298 FB12 9156 Health and Dis
- Page 1851 and 1852:
B12 9154 FVITB 57319 FB12V 7. Benoi
- Page 1853 and 1854:
B6PRO 61064 FBIOT 91902 VITE Vitami
- Page 1855 and 1856:
VLTB 89190 the single most importan
- Page 1857 and 1858:
VLTU 8826 VHLD Toxic concentration:
- Page 1859 and 1860:
VHLSP 89083 Hes FJ, Hoppener JWM, L
- Page 1861 and 1862:
9862 VWD2N 81662 contrast, nonsense
- Page 1863 and 1864:
VWAG 9051 Useful For: Diagnosis of
- Page 1865 and 1866:
VWPR 83099 Inherited vWD has been c
- Page 1867 and 1868:
FVORI 91998 VORI 88698 FPIKE 91662
- Page 1869 and 1870:
WARFP 60529 immune response to alle
- Page 1871 and 1872:
WARFO 60341 impaired. Reference Val
- Page 1873 and 1874:
9943 WSPV 82659 useful for evaluati
- Page 1875 and 1876:
WEED2 81883 immune response to alle
- Page 1877 and 1878:
WEED4 81885 4 17.5-49.9 Strongly po
- Page 1879 and 1880:
WNVP 87802 Clinical Information: We
- Page 1881 and 1882:
WEEPC 83918 WEEP 83156 virus: a pri
- Page 1883 and 1884:
FWHET 91652 FWHT4 91978 sensitizati
- Page 1885 and 1886:
ASHW 82730 sensitization and clinic
- Page 1887 and 1888:
WFHV 82658 WHIC 82719 6 > or =100 S
- Page 1889 and 1890:
POTA 82710 sensitization to particu
- Page 1891 and 1892:
WSLK 82772 sensitivity to inhalant
- Page 1893 and 1894:
WDKM 83698 Willow, IgE Clinical Inf
- Page 1895 and 1896:
FWHS 88535 WORM 82680 2008;47(6):20
- Page 1897 and 1898:
XHIM 82964 present (eg, 45,X/46,XX)
- Page 1899 and 1900:
FBMT 80601 FXYLP 90359 XX/XY in Opp
- Page 1901 and 1902:
YMICR 82992 FYSTG 92000 0-16 years:
- Page 1903 and 1904:
FYERS 57374 ZAP70 83727 clinical ma
- Page 1905 and 1906:
ZNU 8591 ZNRU 60526 lead-poisoning
- Page 1907 and 1908:
ZNCRU 60527 ingestion relates to th
- Page 1909:
FZUCE 92006 ZYG 81252 4. Kawada K,